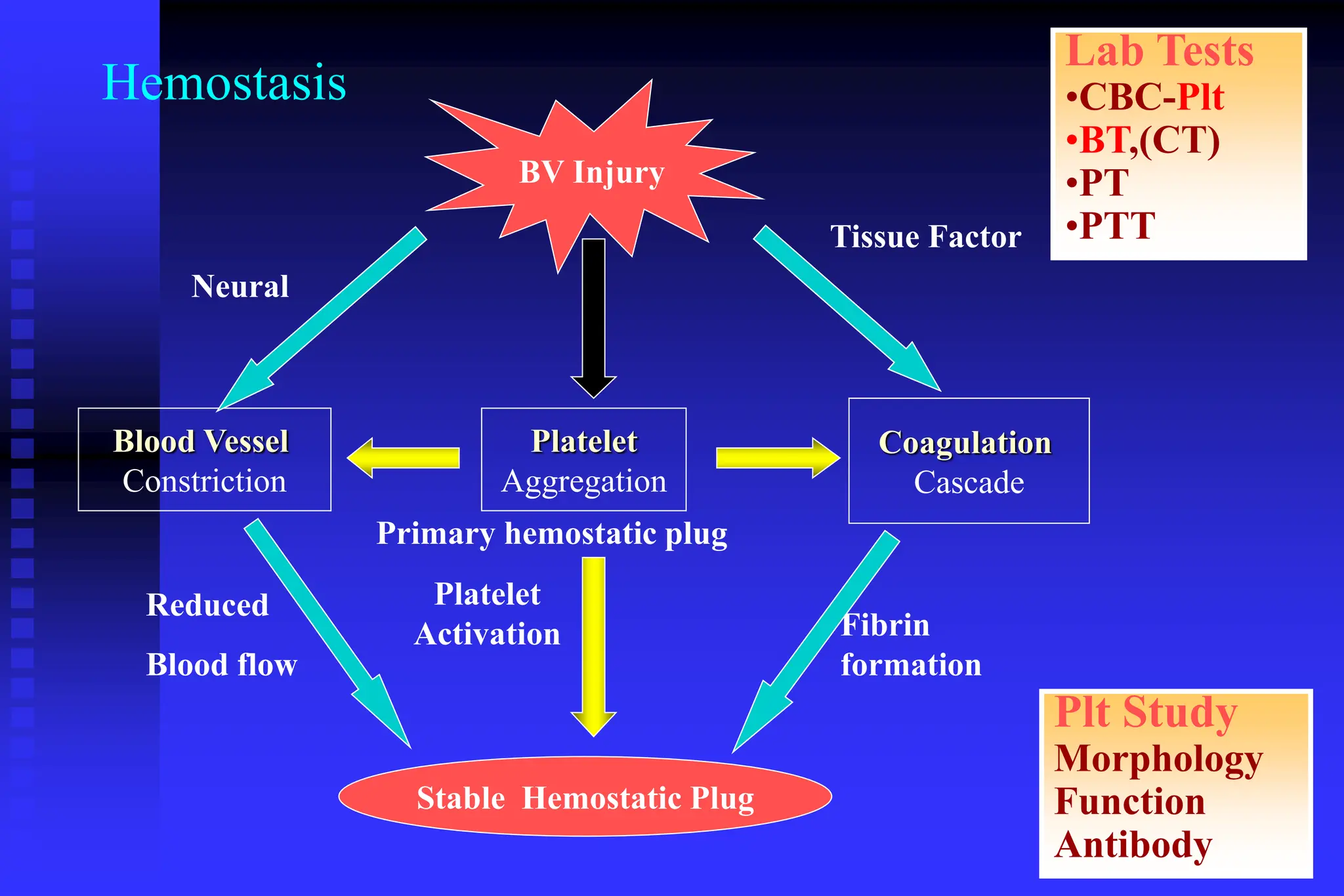
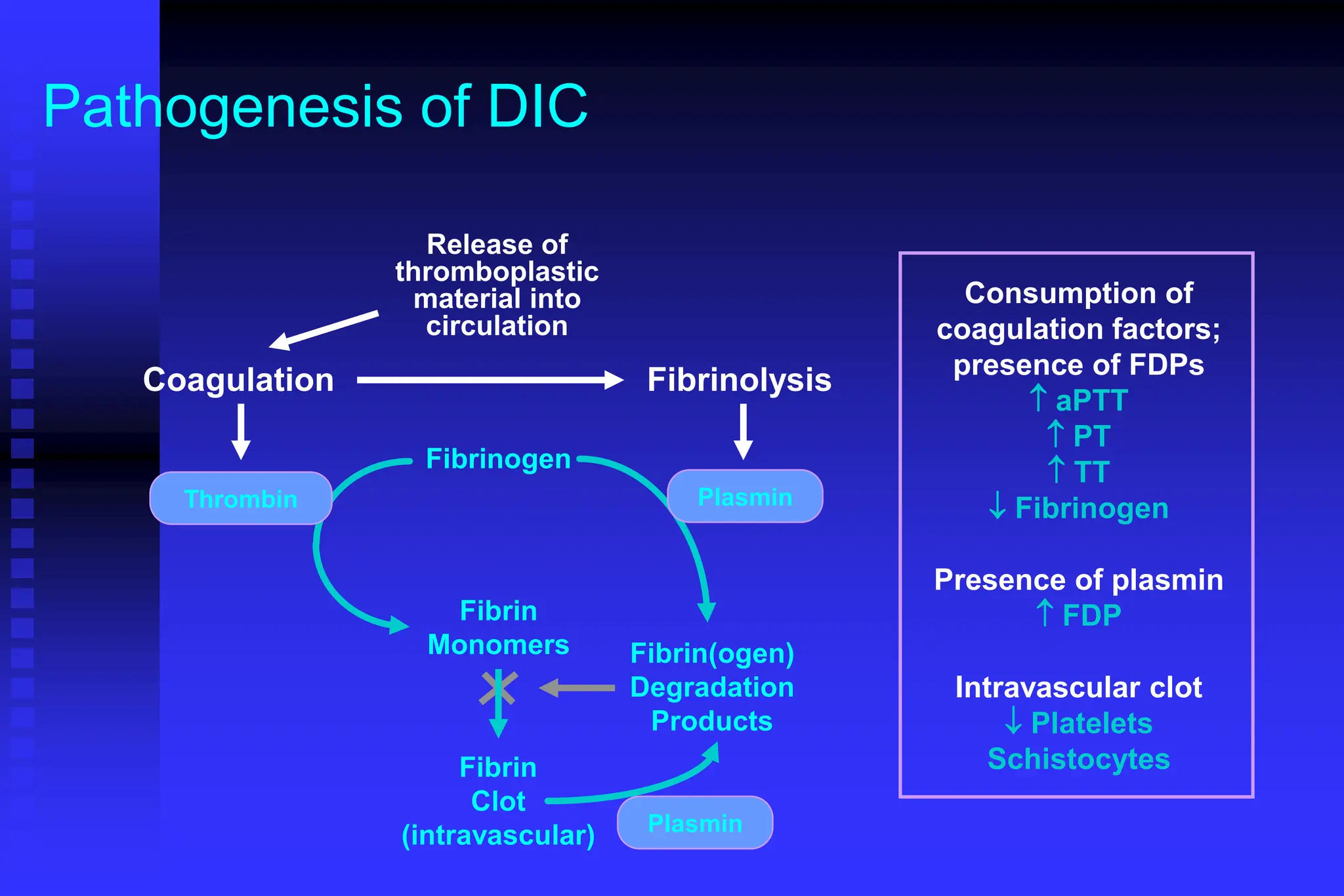
The document outlines the phases and mechanisms of hemostasis, detailing the vascular, platelet, coagulation, and fibrinolytic phases involved in blood clotting. It discusses various bleeding disorders, their causes, laboratory evaluations, and differences in clinical features between platelet and coagulation factor disorders. It also addresses treatment methods, emphasizing the importance of factors such as vessel integrity, adequate platelet levels, and proper function of clotting pathways.