Downloaded 13 times





![FASTQ
Contains 3 things per read:
- Sequence identifier (unique)
- Sequence bases [len N]
- Base quality scores [len N]
Often “gzip” compressed (fq.gz)
If not demultiplexed, first 4 or 6bp
is the “barcode” index. Used to
split lanes out by sample.
Filtering may include:
- Removing adapters & primers
- Clip poor quality bases at ends
- Remove flagged low-quality reads
@HWI-ST845:4:1101:16436:2254#0/1
CAAACAGGCATGCGAGGTGCCTTTGGAAAGCCCCAGGGCACTGTGGCCAG
+
Y[SQORPMPYRSNP_][_babBBBBBBBBBBBBBBBBBBBBBBBBBB](https://image.slidesharecdn.com/cslecture2017-04-11fromdatatoprecisionmedicine-170411234349/75/CS-Lecture-2017-04-11-from-Data-to-Precision-Medicine-12-2048.jpg)







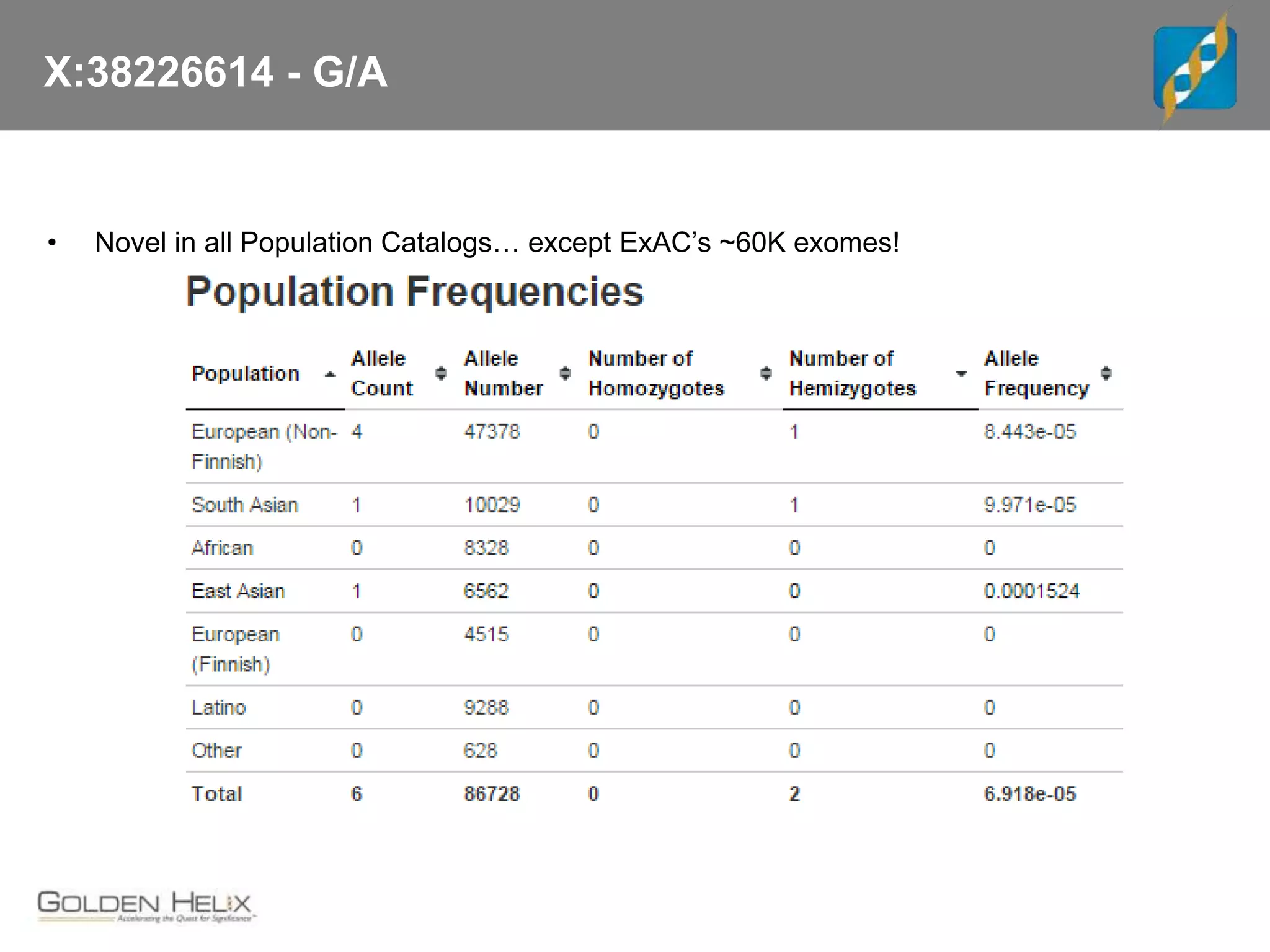

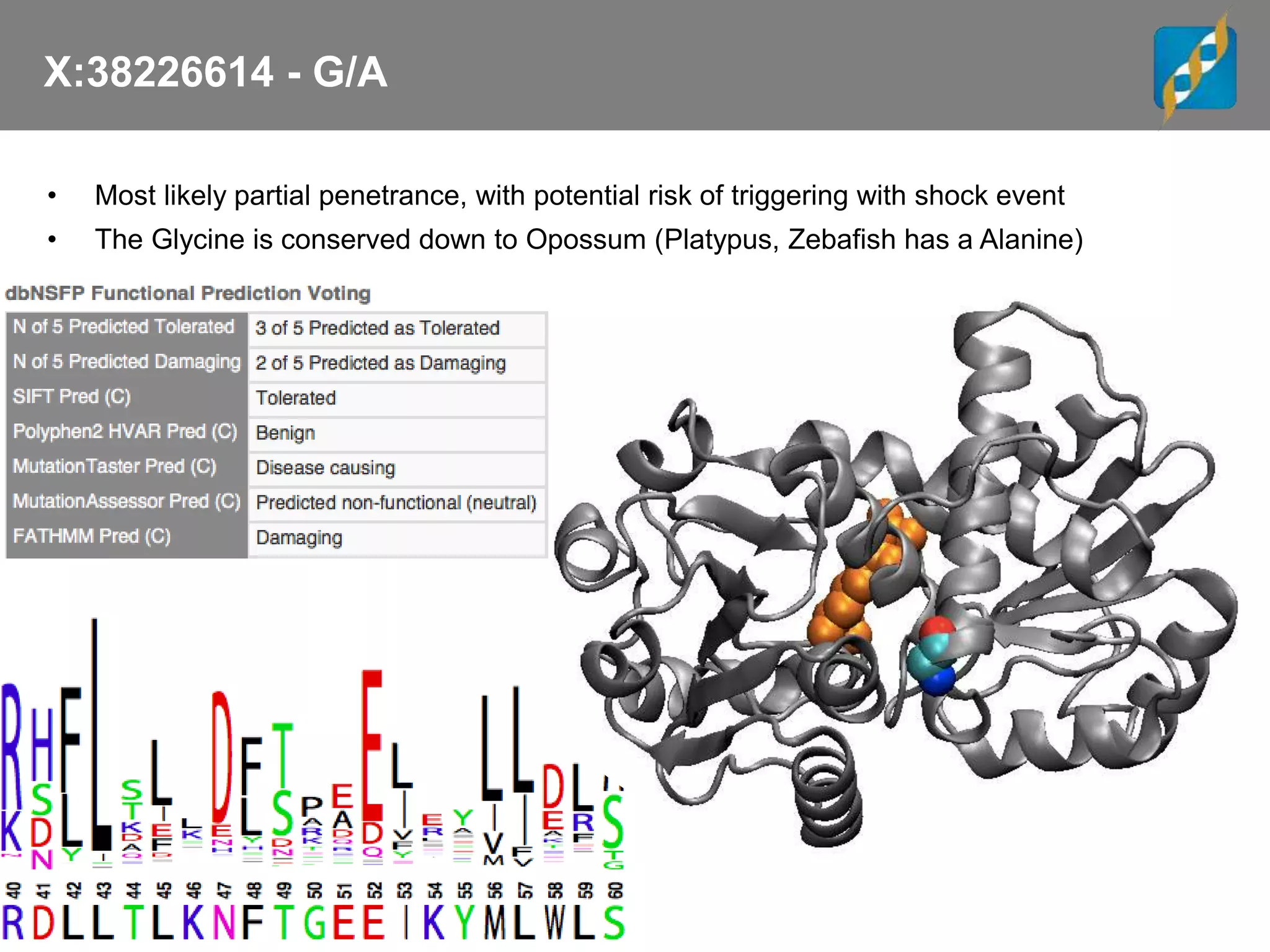
![X:38226614 - G/A
• Cited PubMed article was on ResearchGate, Hiroki Morizono contacted
• Provided full text and lots of interesting backstory on OTC
• “If you are able to eat all the steak you want, you may have the mutation; it would
appear to be a hypomorphic allele (and a very mild one at that)”
• “Is possible that the late onset case that [was] identified may have been someone
who was having a very bad day, and several things went poorly for them.”
• “The R40H mutation, there was a grandfather or granduncle who was affected who
ate whatever he wanted, and seemed unaffected while the proband had several
episodes.”](https://image.slidesharecdn.com/cslecture2017-04-11fromdatatoprecisionmedicine-170411234349/75/CS-Lecture-2017-04-11-from-Data-to-Precision-Medicine-28-2048.jpg)


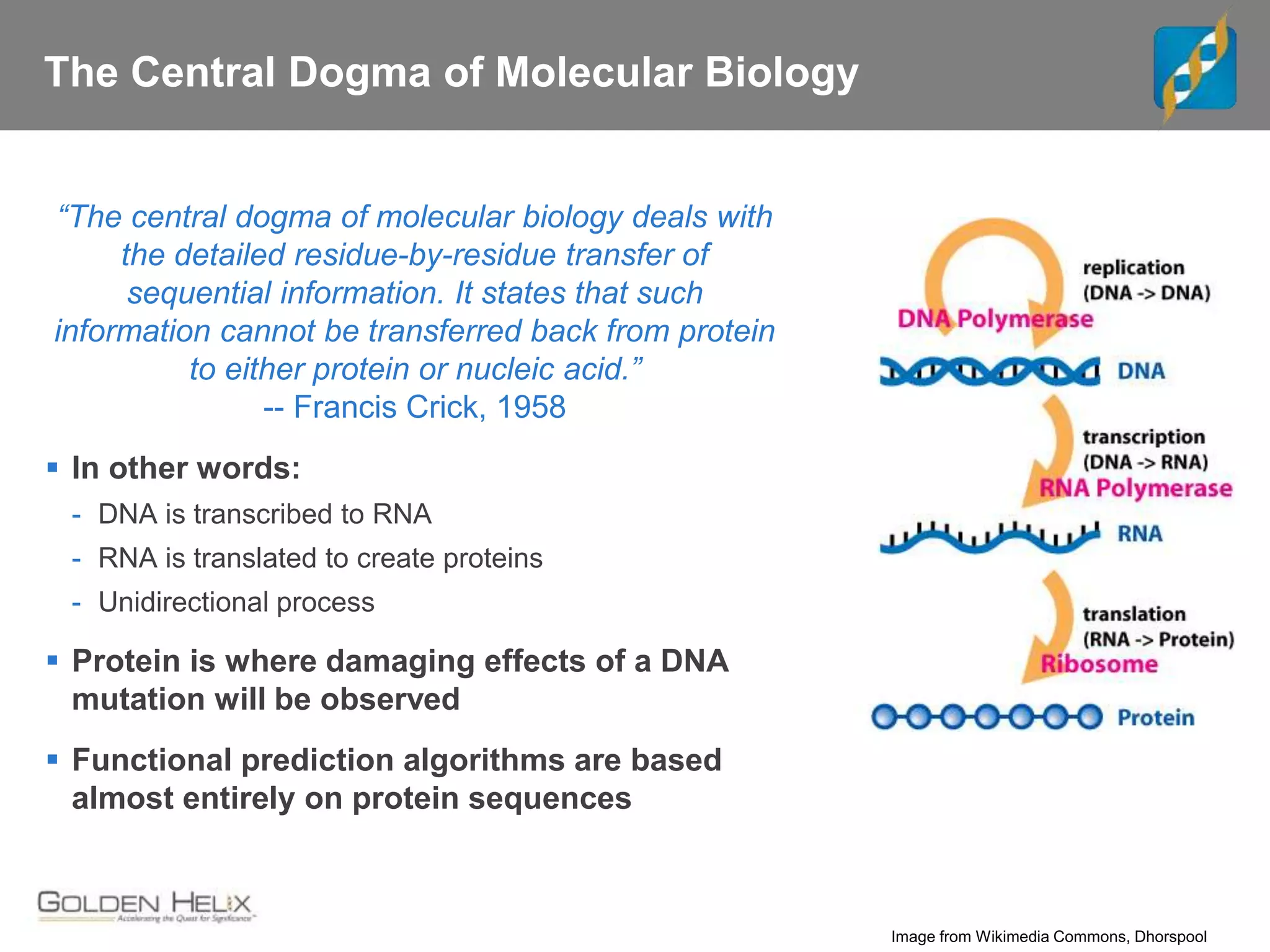
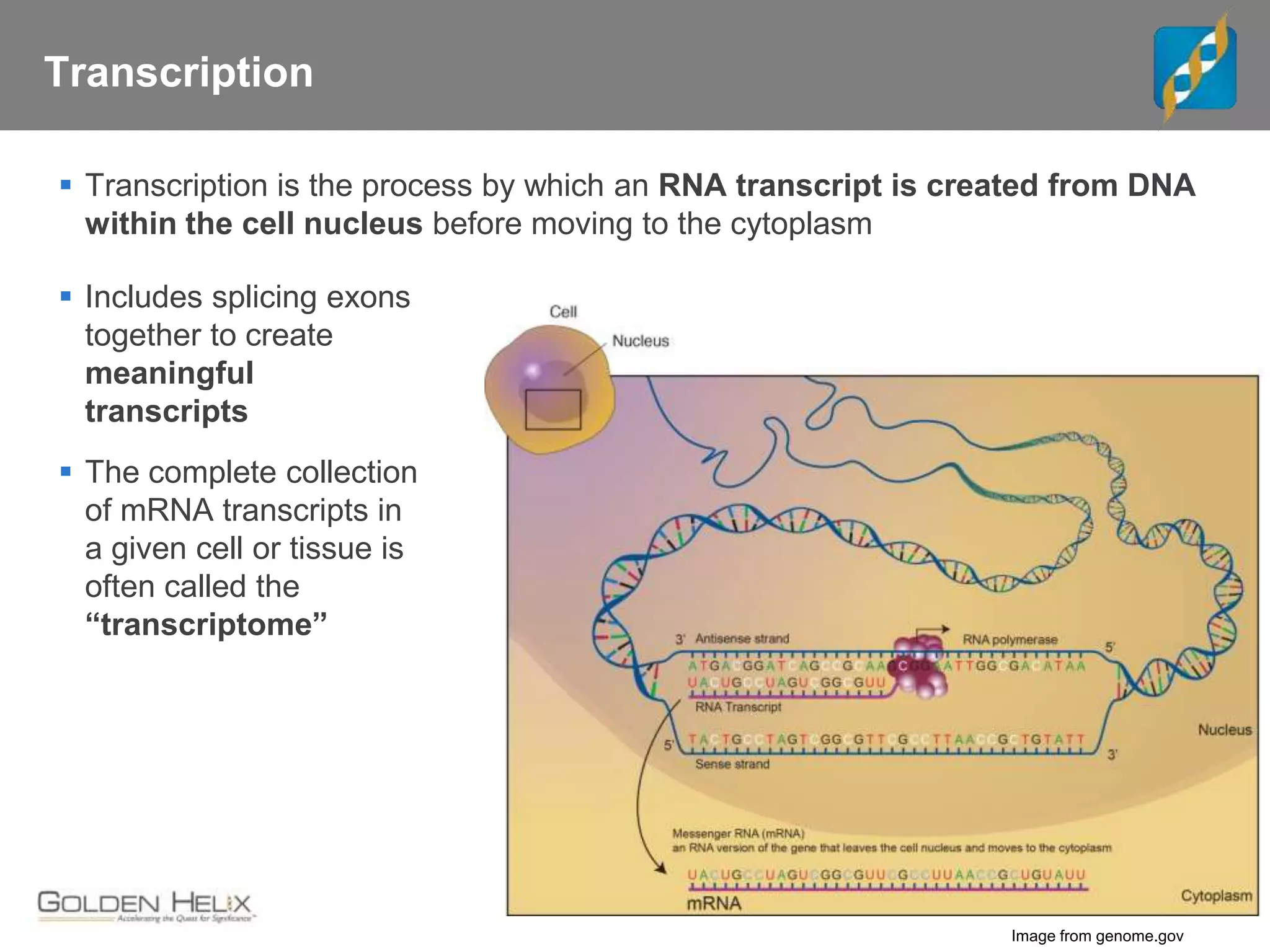
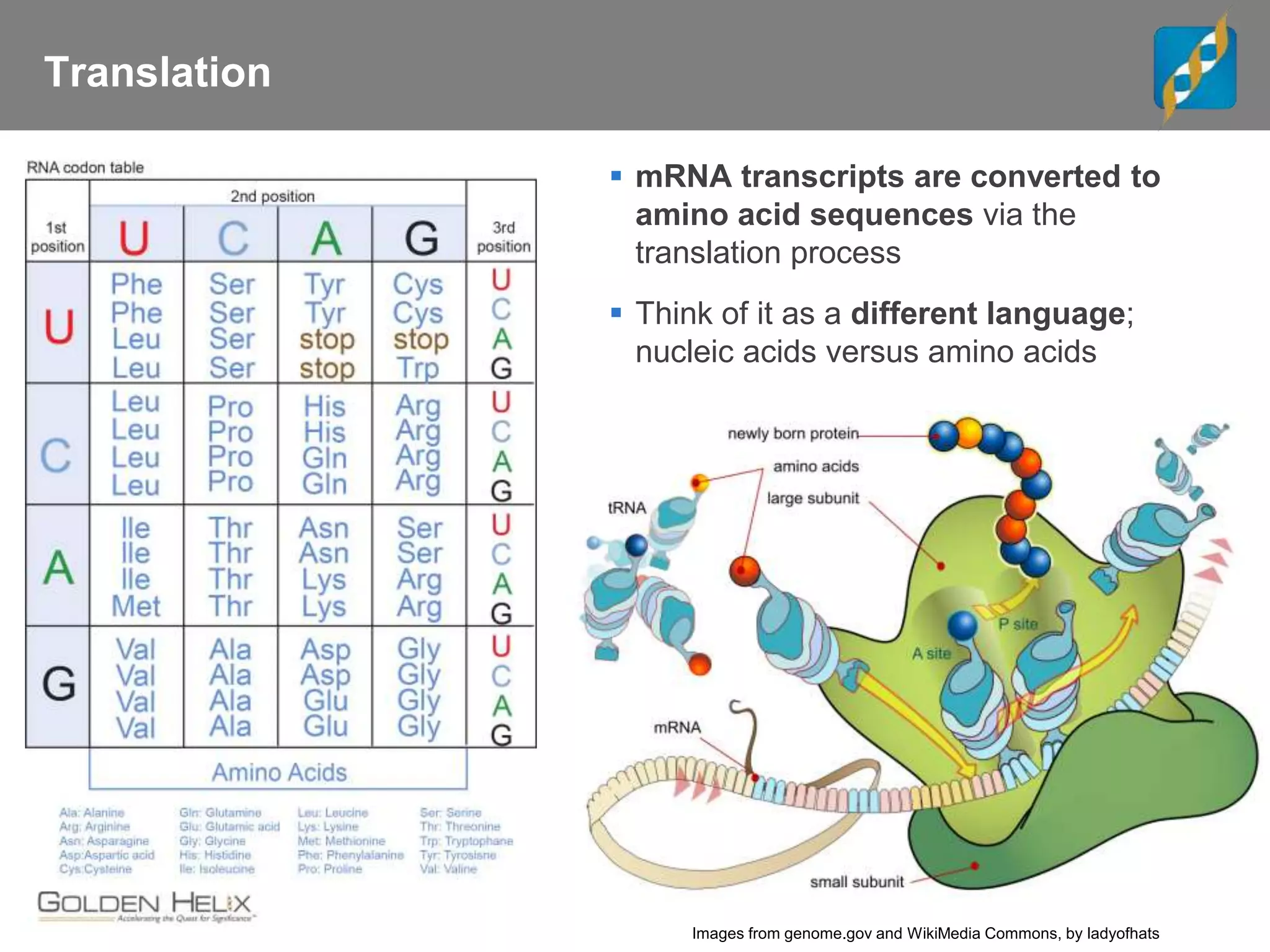
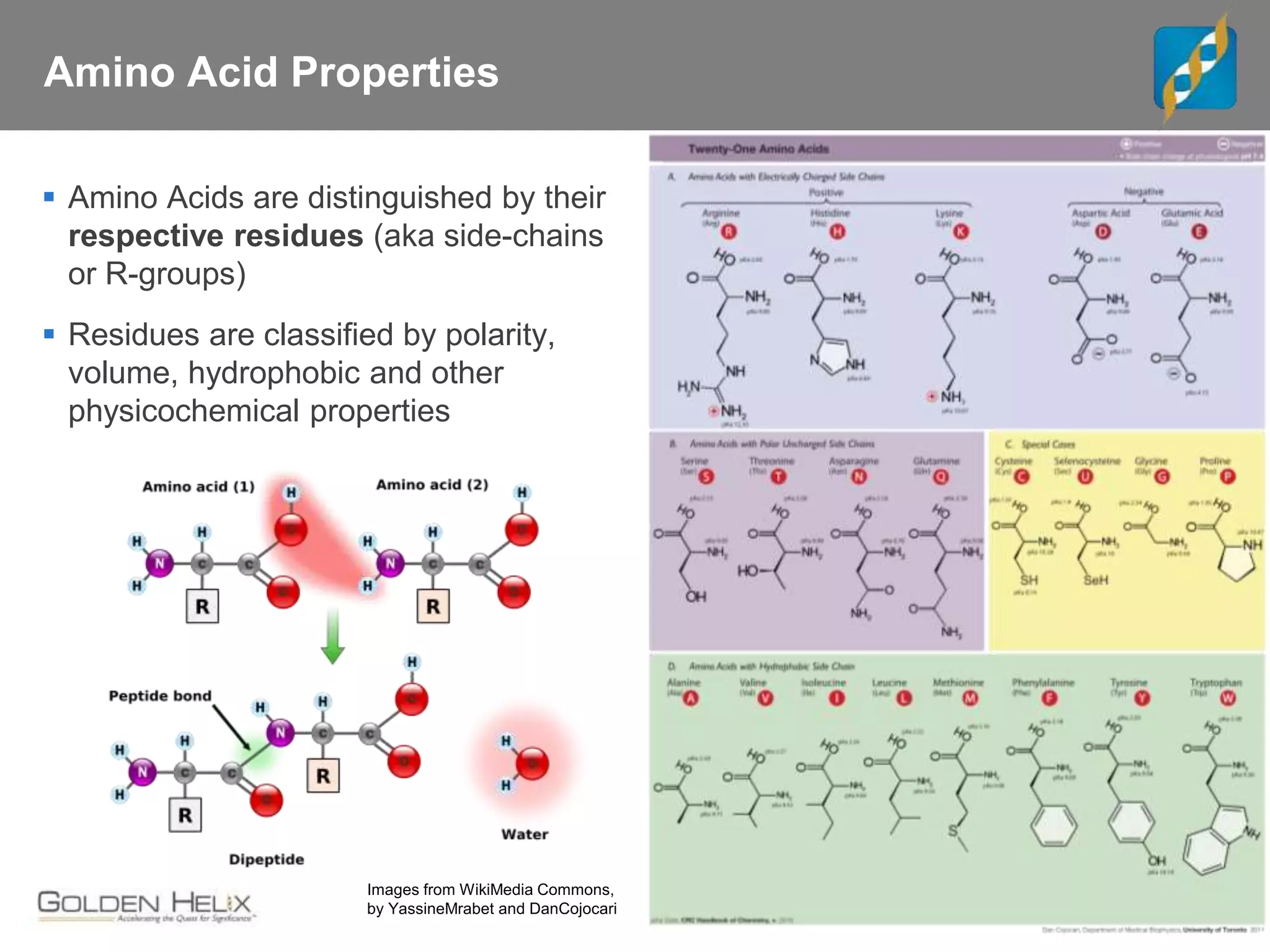
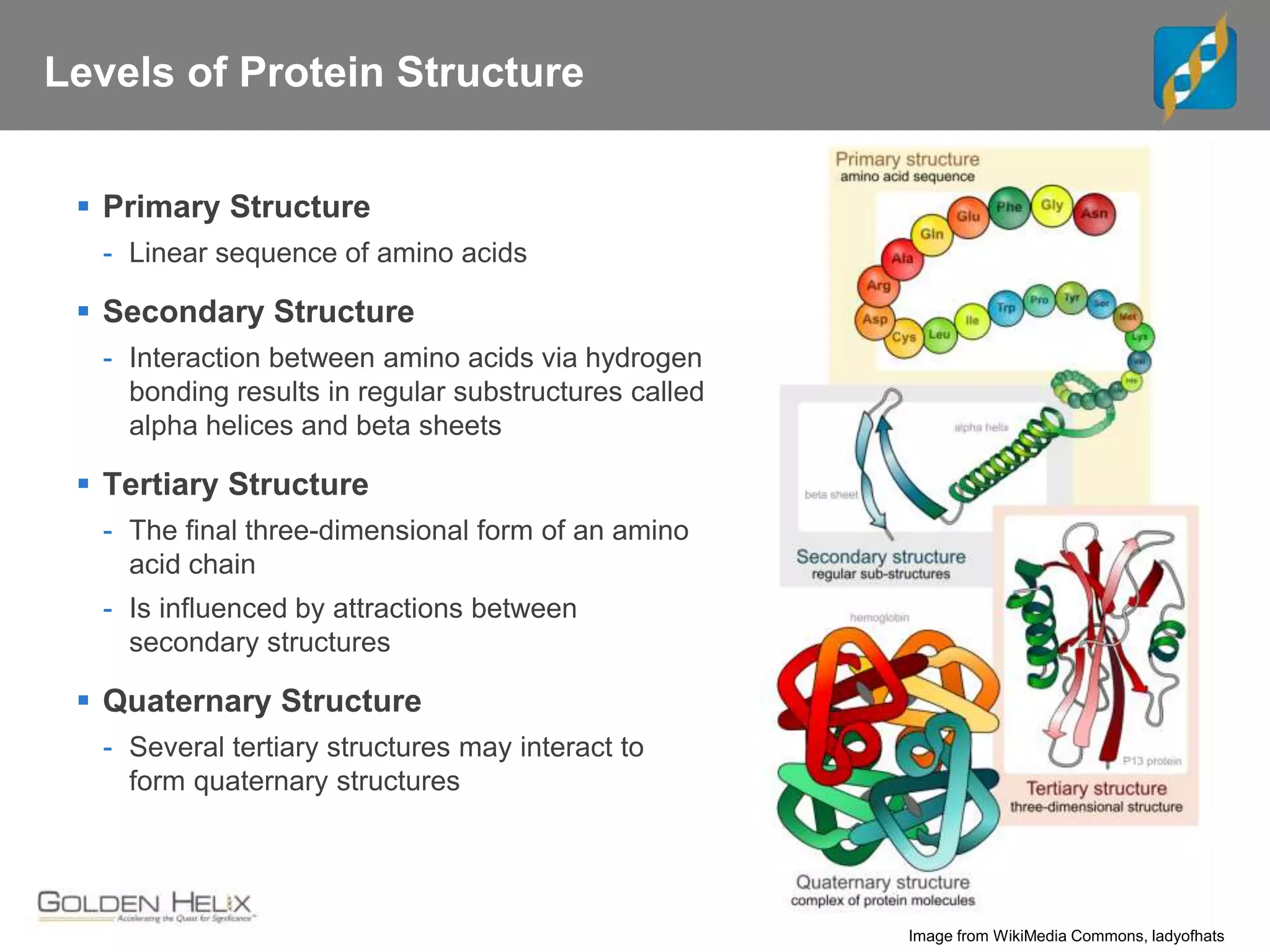
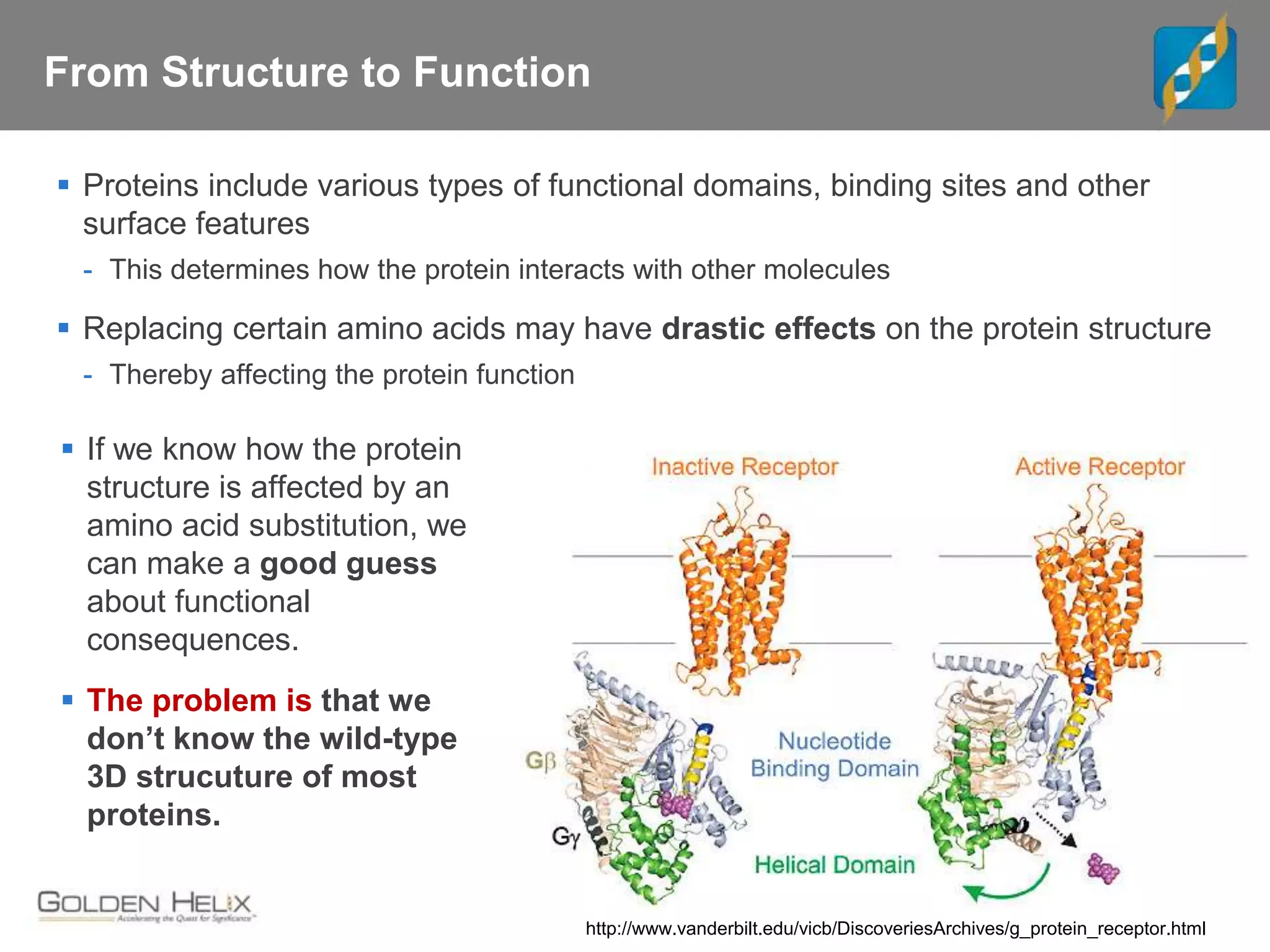


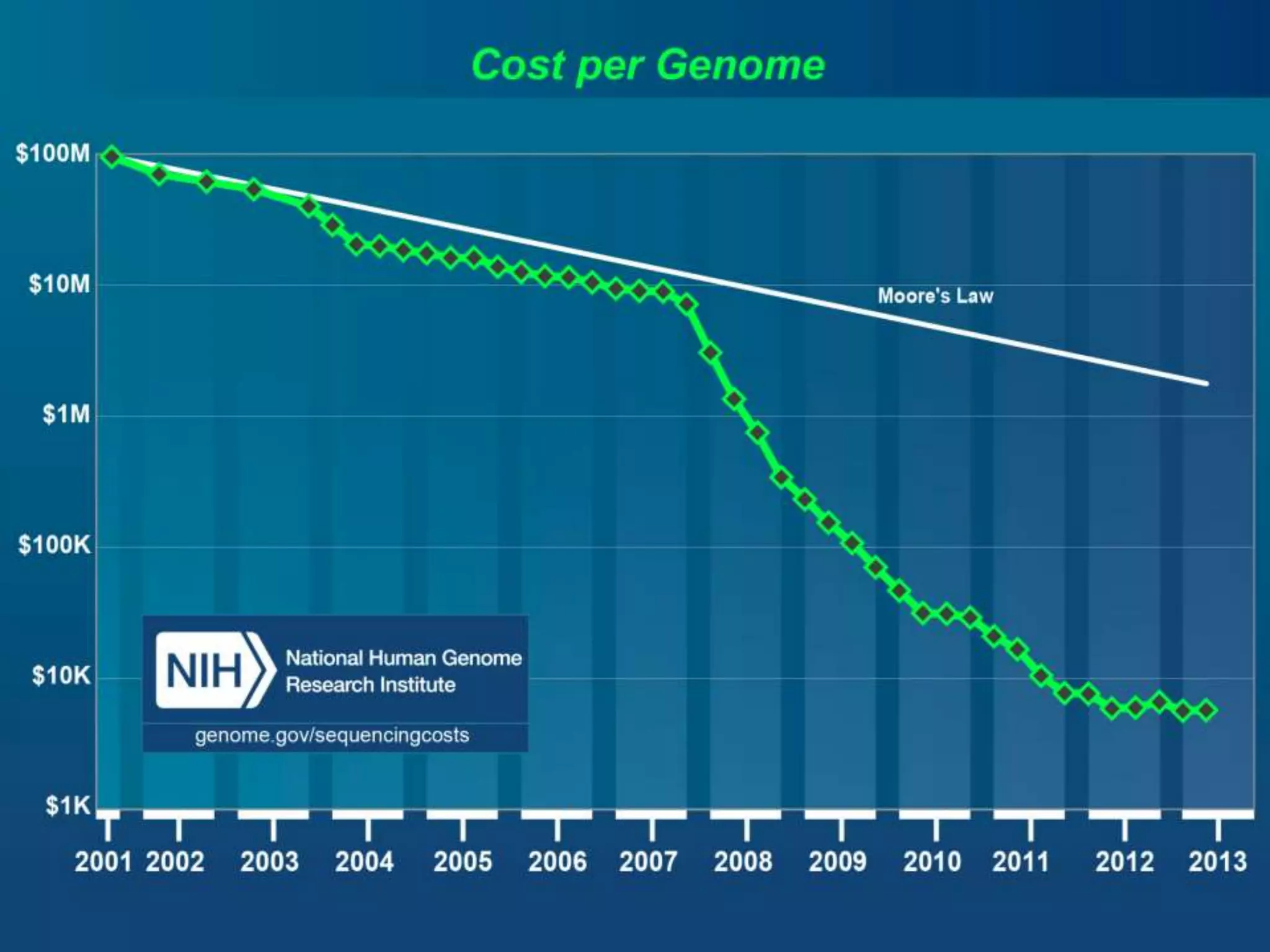
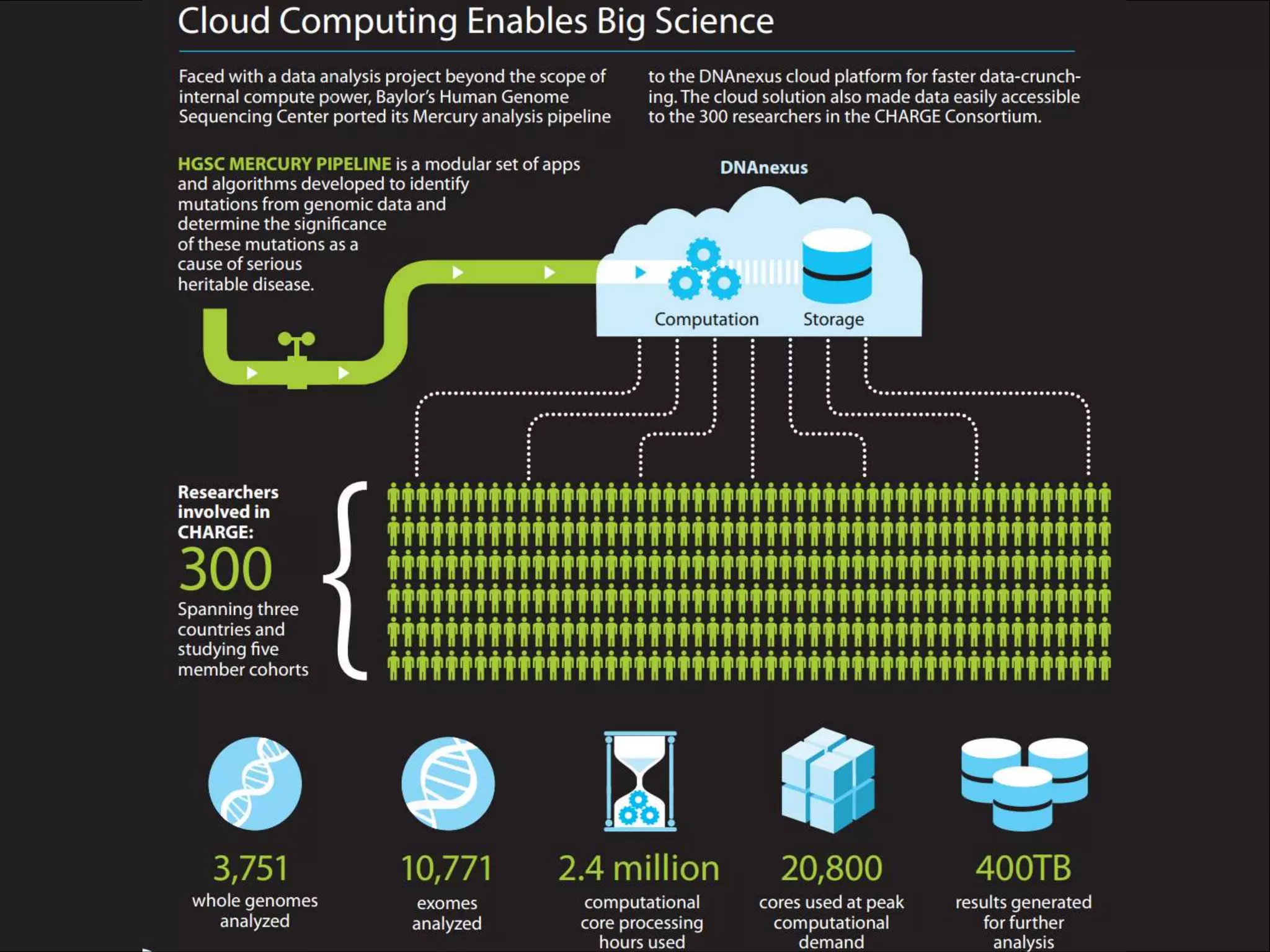
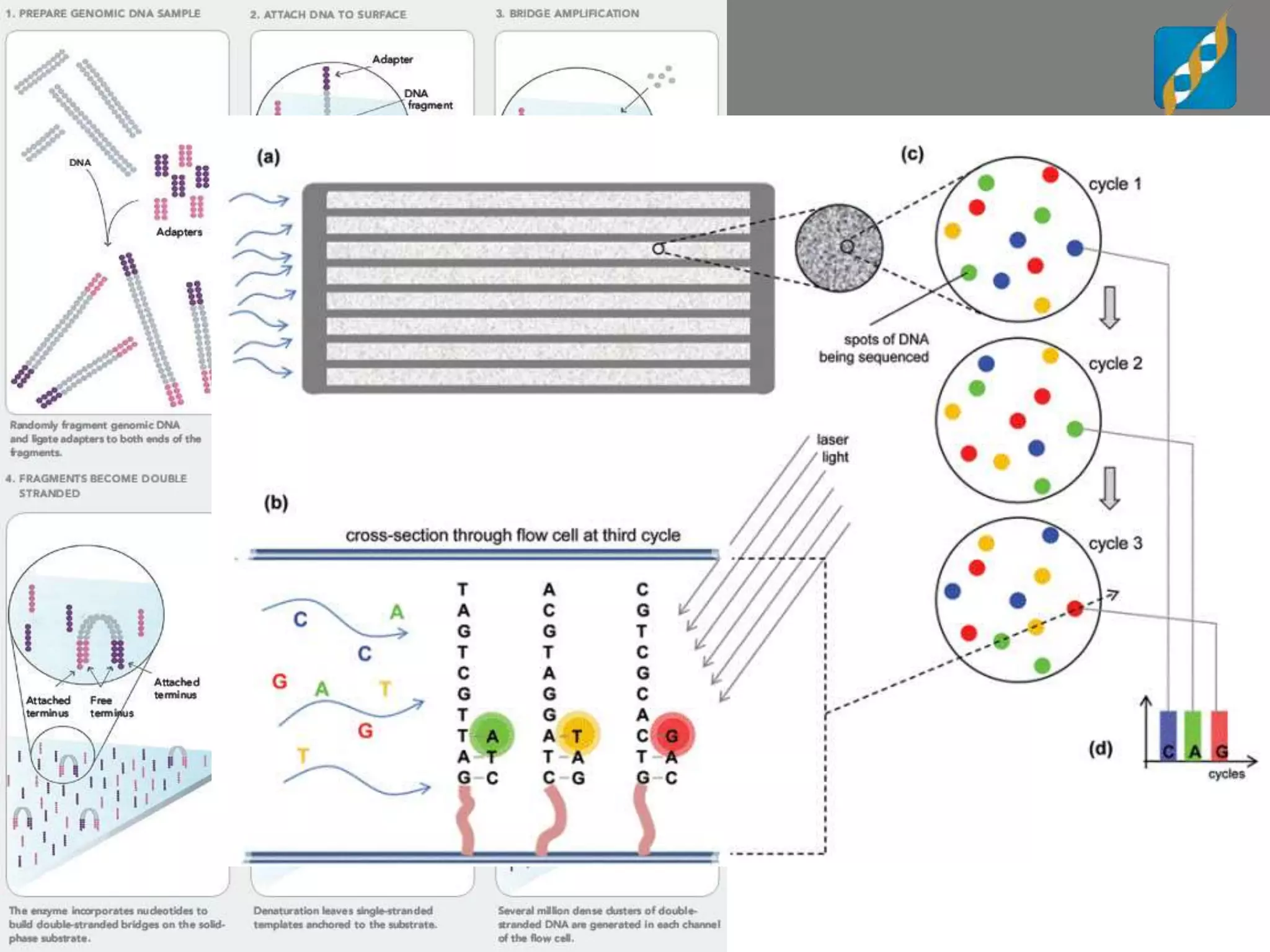
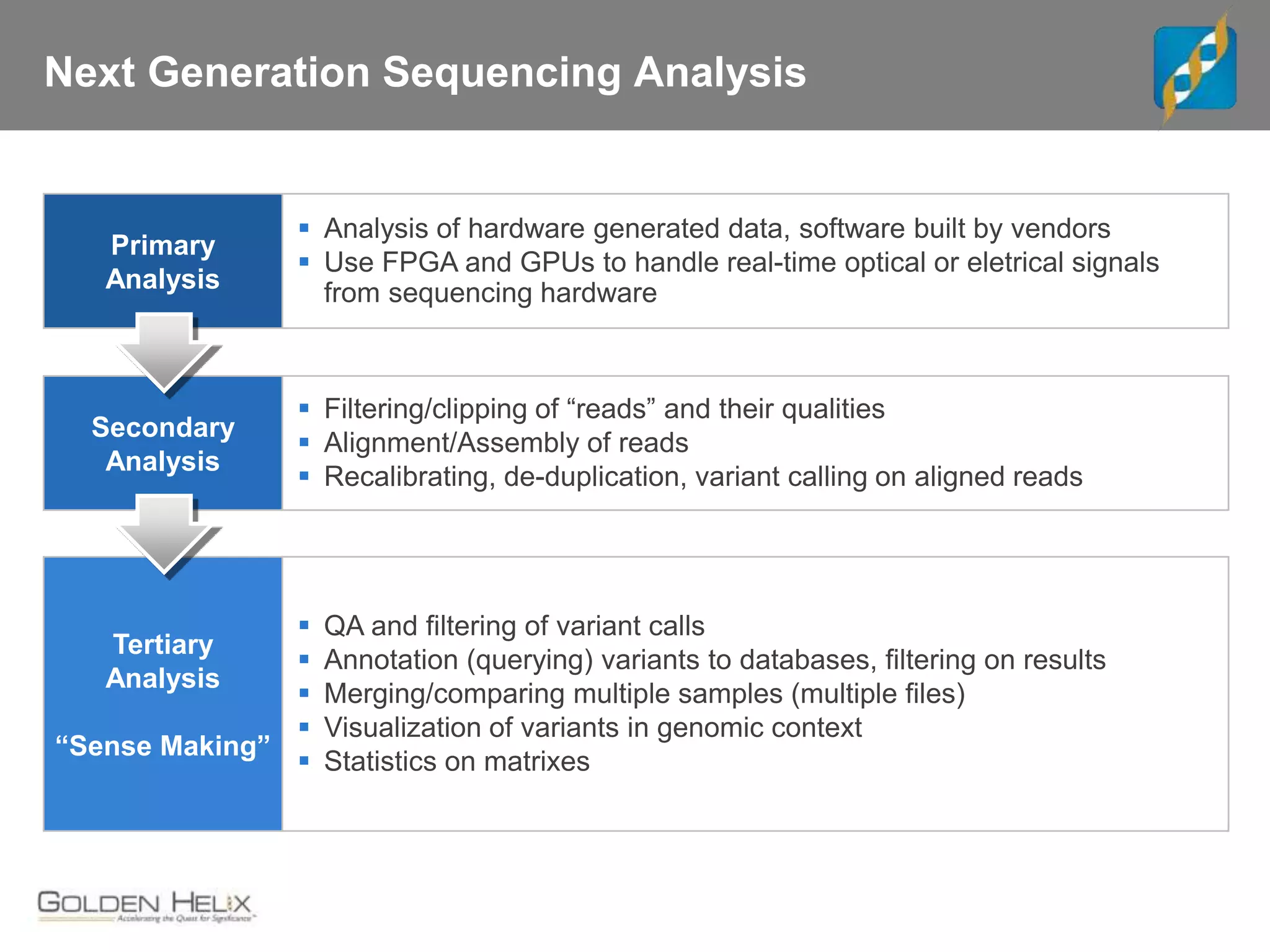
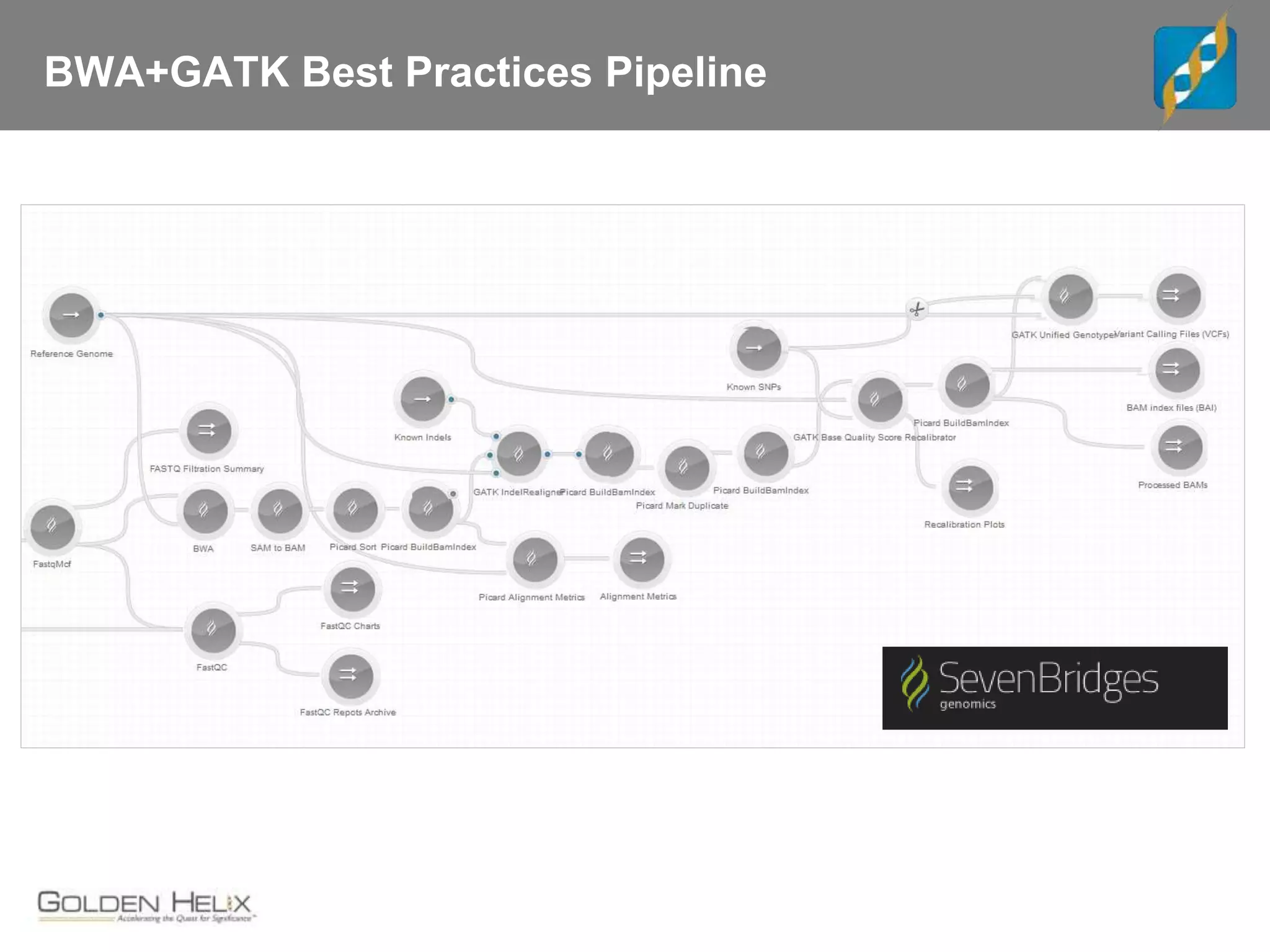
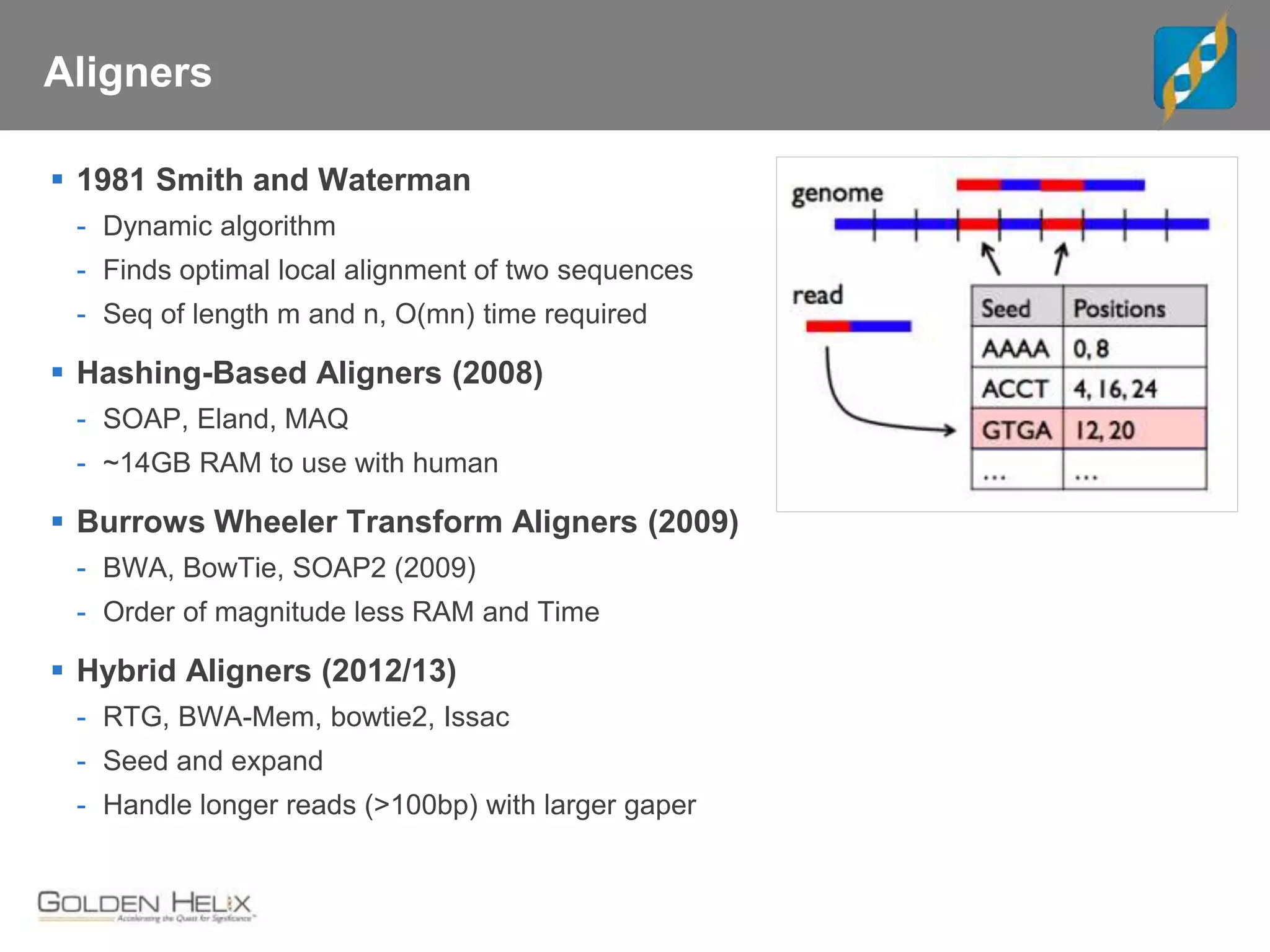
Next Generation Sequencing (NGS) data analysis involves multiple complex steps from raw data generation to interpretation. Primary analysis converts raw instrument files to sequence reads. Secondary analysis aligns reads to a reference and identifies variants. Tertiary analysis annotates variants, filters data, and prioritizes candidates for further study. Key file formats at each step include FASTQ, SAM/BAM, and VCF. Visualization and population databases aid in data interpretation and clinical relevance. The central dogma of molecular biology relates how genetic changes impact the genome, transcriptome and proteome.






















![[DSC Europe 23][DigiHealth] Vesna Pajic - Machine Learning Techniques for omi...](https://cdn.slidesharecdn.com/ss_thumbnails/dxgiw6wysauhxm3dnays-vesna-pajic-machine-learning-techniques-for-omics-data-analysis-231130112724-c1268146-thumbnail.jpg?width=640&height=640&fit=bounds)
























