



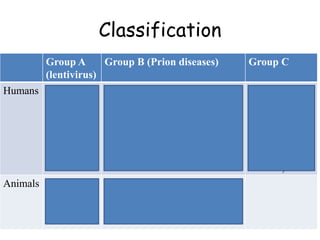






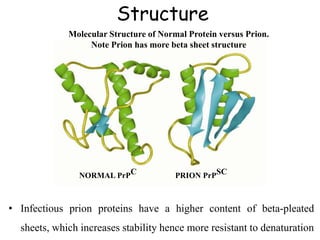
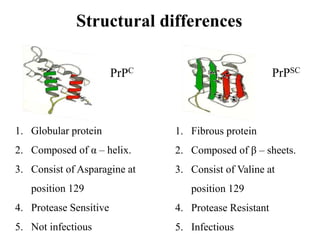
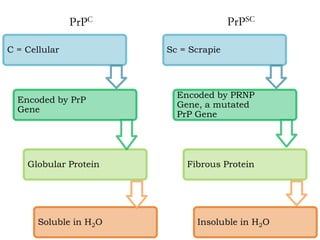
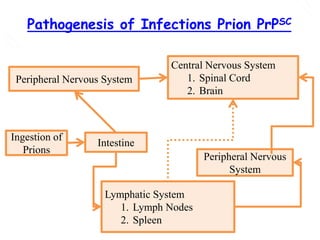

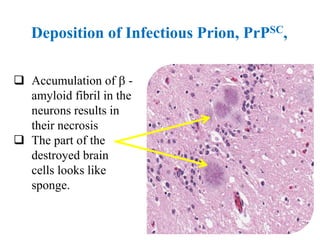




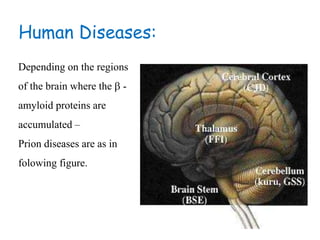









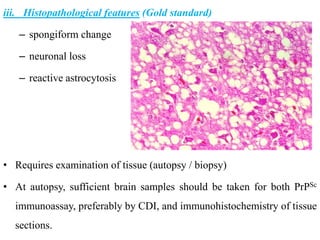











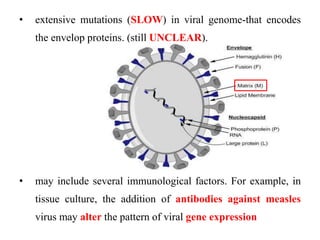






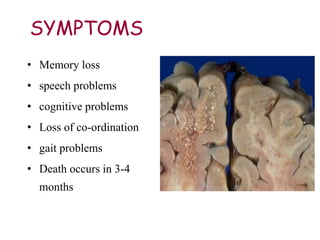
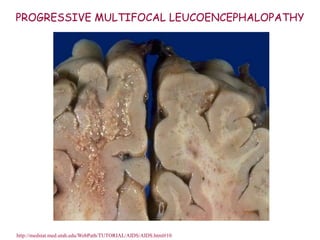

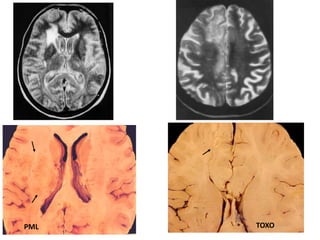


The document discusses slow virus diseases and prion diseases. It describes how Björn Sigurðsson first introduced the slow virus concept and studied diseases like maedi, visna, and scrapie in sheep. These diseases have long incubation periods and involve the central nervous system. Prion diseases like Kuru, Creutzfeldt-Jakob disease, and mad cow disease are also described, noting they involve misfolded prion proteins and have characteristics like spongiform changes in the brain. Measles virus diseases like subacute sclerosing panencephalitis are also slow virus diseases that occur due to mutations in the measles virus.





































![Colistin wc500146813[1]](https://cdn.slidesharecdn.com/ss_thumbnails/colistinwc5001468131-160512033728-thumbnail.jpg?width=640&height=640&fit=bounds)











![Prions_Presentation[1] [Autosaved]..pptx](https://cdn.slidesharecdn.com/ss_thumbnails/prionspresentation1autosaved-250425151925-c892db5b-thumbnail.jpg?width=640&height=640&fit=bounds)
















































