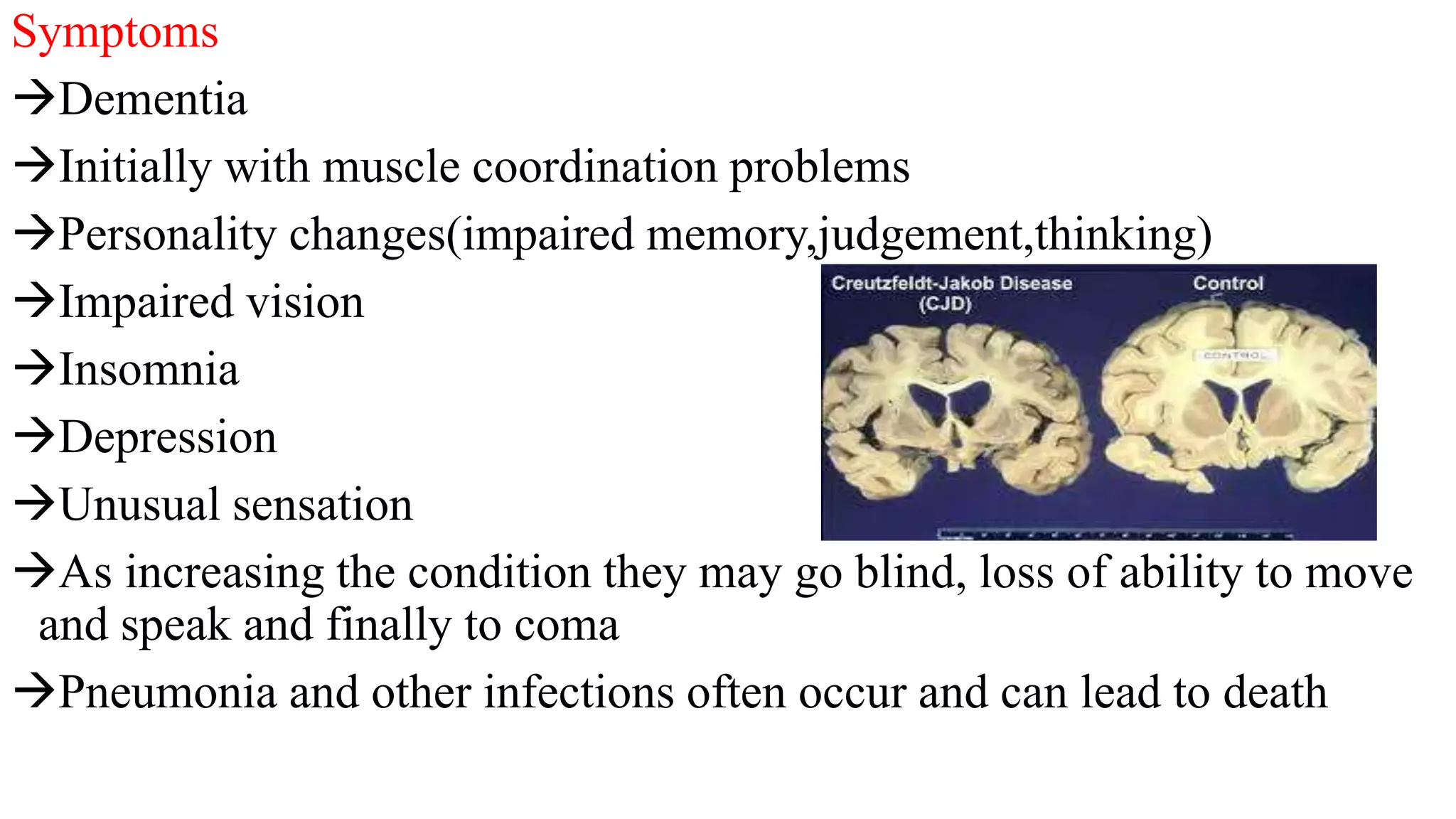
Scrapie is a fatal, neurological disease that affects sheep and goats. It is caused by prions, which are misfolded proteins, and results in the accumulation of prions in the nervous system, leading to neuronal death. Symptoms include weight loss, behavioral changes, and tremors. It is transmitted from infected females to offspring at birth or through contact with birth materials. There is no treatment for scrapie. Control methods focus on selective breeding of resistant sheep genotypes and restricting the spread of infected tissues and milk.








![• Creutzfeldt–Jakob disease (CJD), also known as subacute
spongiform encephalopathy or neurocognitive disorder due to prion
disease, is a fatal degenerative brain disorder
• CJD is caused by a protein known as a prion.[5] Infectious prions
are misfolded proteins that can cause normally folded proteins to
become misfolded
• It is a very rare fatal neurodenerative syndrome that causes 1 in million
• Depending on the reason of appearance it is categorized in to different
types they are
• Sporadic CJD appears even though there are no risk factors
• Heriditary CJD from family members
• Aquired CJD exposure to brain through medical procedures or eating
meat of containg infected prions](https://image.slidesharecdn.com/scrapieandcjd-210405152216/75/prion-diseases-Scrapie-and-cjd-9-2048.jpg)