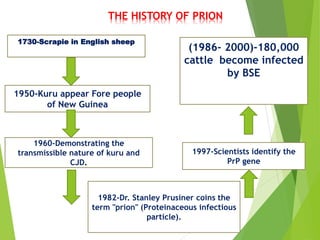
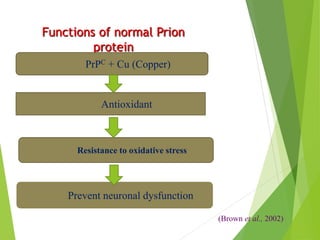
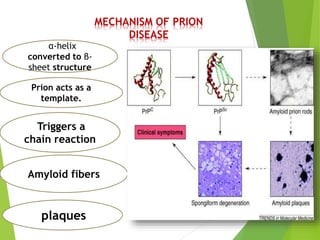
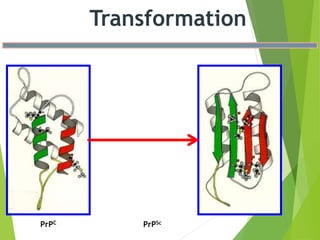
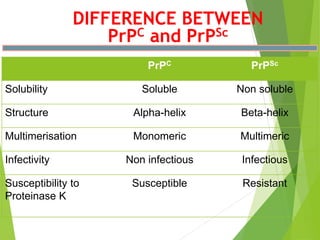
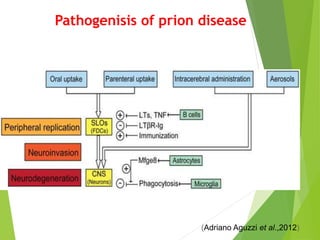
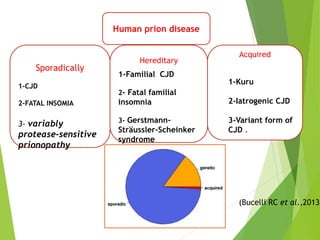
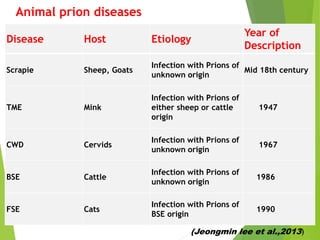
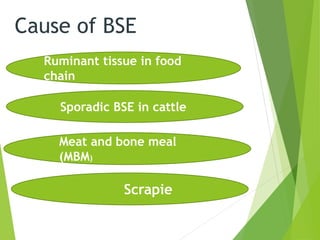
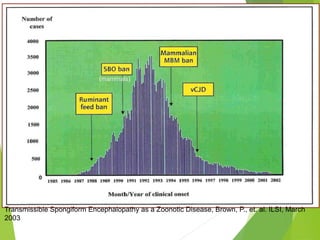
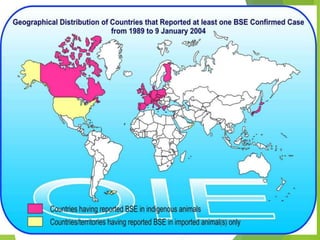
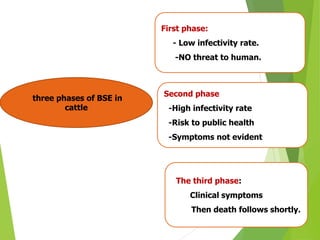
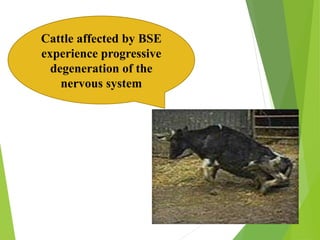
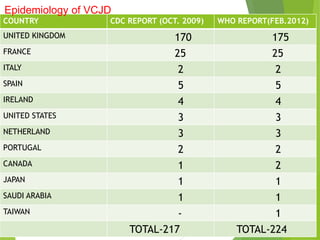
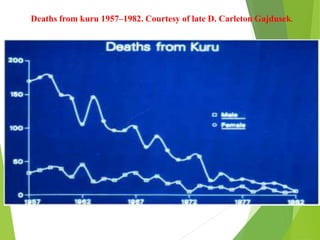
The document provides a comprehensive overview of prion diseases, detailing their history, pathogenesis, and various forms in both humans and animals. It emphasizes the role of misfolded prion proteins in neurodegenerative diseases like Creutzfeldt-Jakob disease and Bovine Spongiform Encephalopathy, highlighting their transmissible nature and fatal consequences. The document also discusses diagnostic methods and control measures to mitigate prion disease outbreaks.