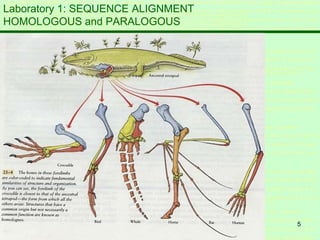
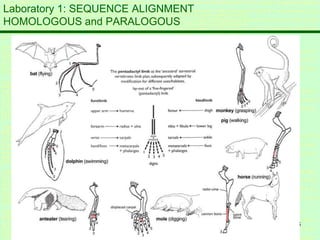
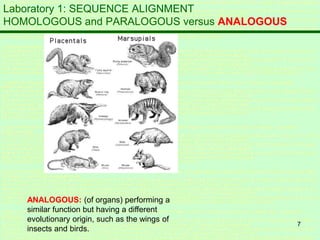
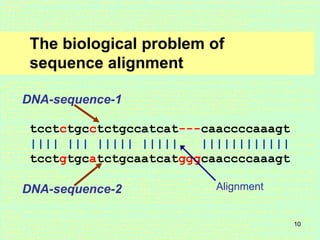
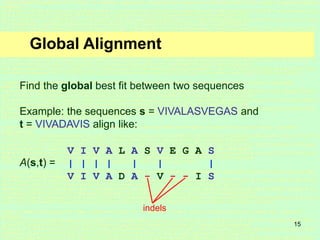
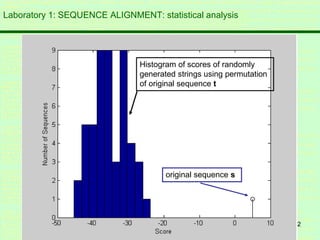
This document discusses sequence alignment, which is important for predicting function, database searching, gene finding, and studying sequence divergence. It describes global and local alignment, and algorithms like Needleman-Wunsch, Smith-Waterman, and BLAST that are used for sequence alignment. Sequence alignment finds the best match between sequences and can provide information about molecular evolution by identifying mutations, insertions, and deletions.