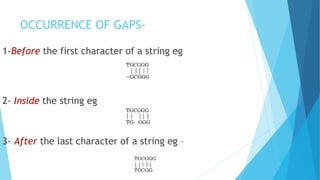
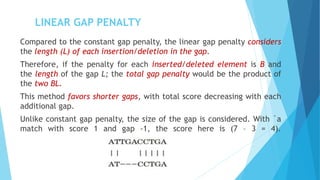
This document provides an introduction to sequence alignment and discusses gaps and gap penalties. It defines a match and gap in sequence alignment and how substitutions, deletions and insertions are represented. It describes different types of gaps including constant, linear, affine, convex and profile-based variable penalties. Highlights include that gaps allow alignment extension and introduce uncertainty, so penalties are used. Examples demonstrate assigning regular and affine gap penalties.