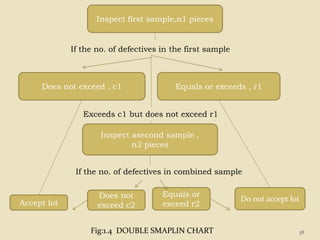
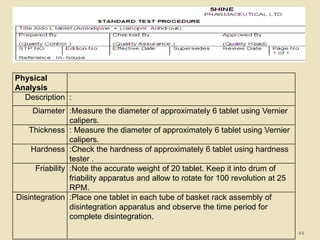
This document provides an overview of quality control procedures and responsibilities in a pharmaceutical quality control laboratory. It discusses the roles of the laboratory director and technicians. It describes routine control instruments, reagent handling and labeling procedures, and various sampling plans including single, double, continuous, and sequential sampling. Standard test procedures and documentation requirements are also outlined. The document establishes best practices for ensuring accurate and reliable testing in the quality control laboratory.















































































































![Good Laboratory Practice (GLP) - General [Manual]](https://cdn.slidesharecdn.com/ss_thumbnails/goodlaboratorypracticeglp-generalmanual-fouadashraf-210330110045-thumbnail.jpg?width=640&height=640&fit=bounds)




























