Downloaded 109 times








































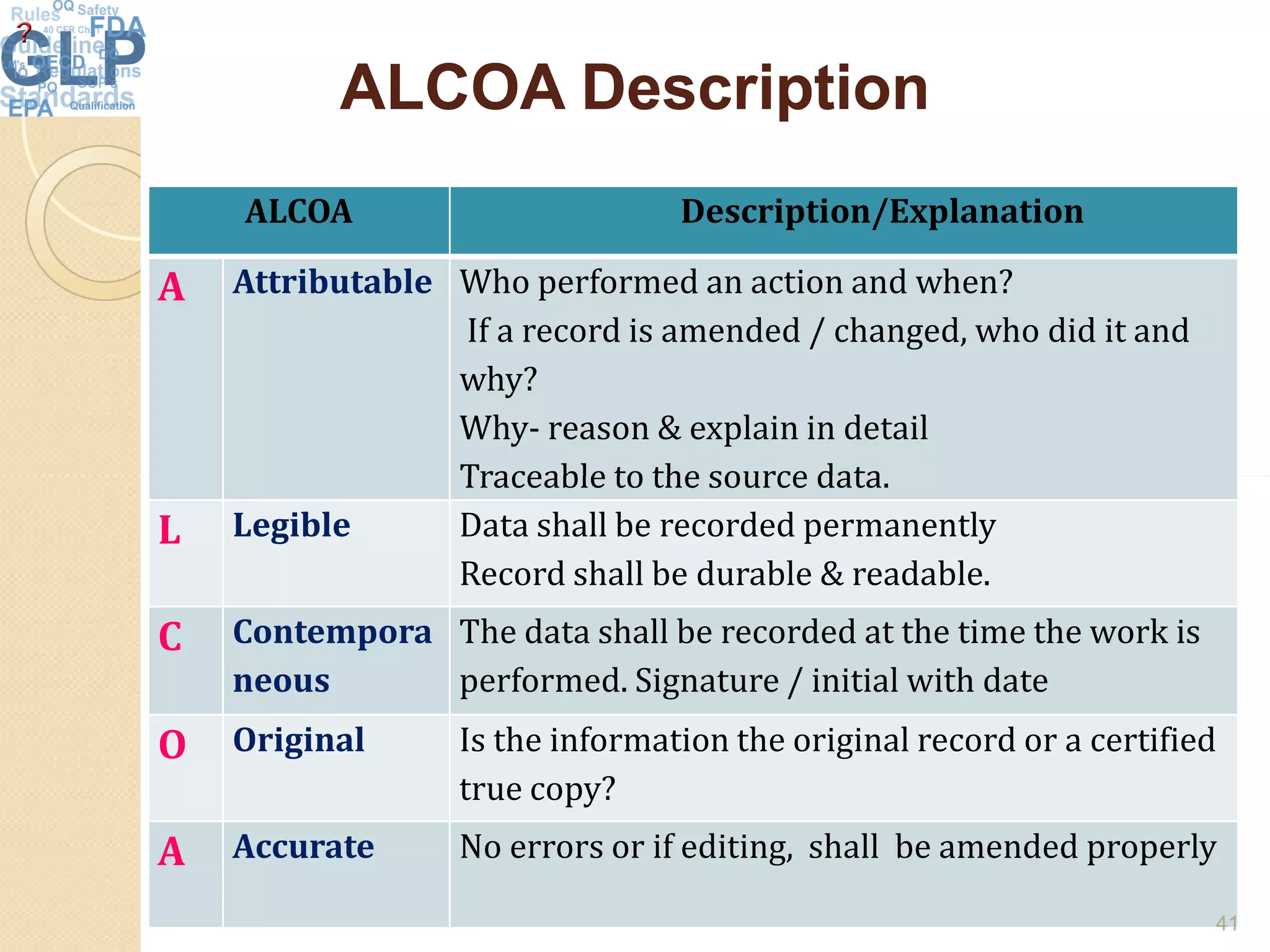
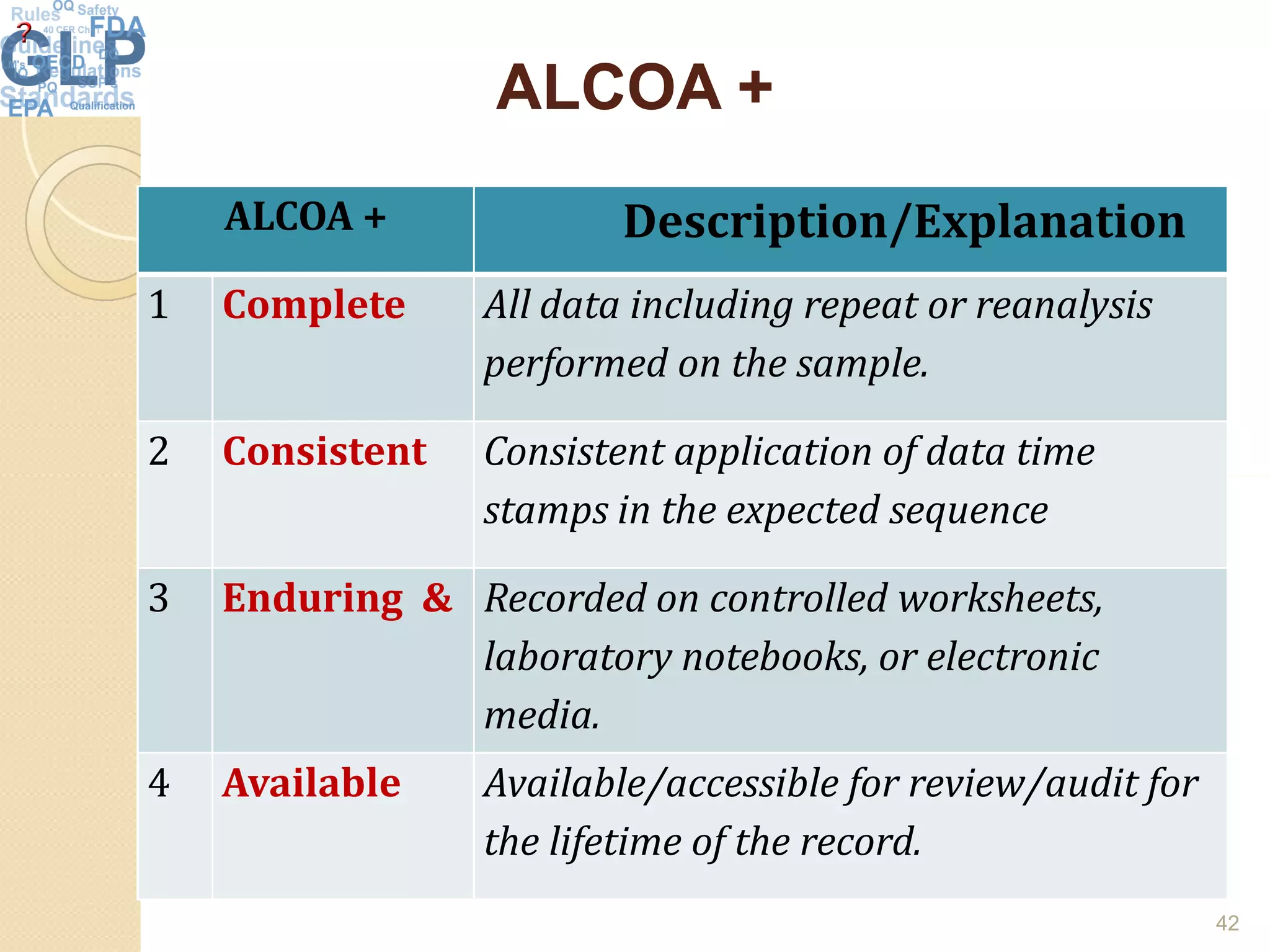












The document outlines good laboratory practices (GLP) in pharmaceutical quality control, emphasizing compliance with regulatory guidelines such as ICH Q7 and 21 CFR 210/211. Key areas covered include laboratory management, personnel qualifications, documentation and record-keeping, sample management, reagents, instruments, and validation processes. The document serves as a comprehensive framework for ensuring quality control and integrity in laboratory operations.
































![Good Laboratory Practice (GLP) - General [presentaion]](https://cdn.slidesharecdn.com/ss_thumbnails/goodlaboratorypracticepresentaion2-210406103432-thumbnail.jpg?width=640&height=640&fit=bounds)
![Good Laboratory Practice (GLP) - General [Manual]](https://cdn.slidesharecdn.com/ss_thumbnails/goodlaboratorypracticeglp-generalmanual-fouadashraf-210330110045-thumbnail.jpg?width=640&height=640&fit=bounds)





















































