Downloaded 316 times

![URS [User Requirement Specification]
DQ [Design Qualification]
IQ [Installation Qualification]
OQ [Operation Qualification]
PQ [Performance Qualification]](https://image.slidesharecdn.com/qualification-150402043319-conversion-gate01/85/Qualification-2-320.jpg)
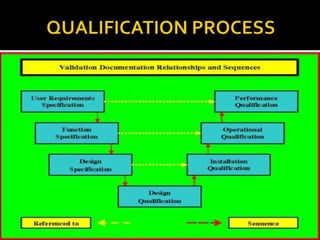






















This document provides information on various qualification documents used in pharmaceutical industries, including: - User Requirement Specification (URS) which documents the end user requirements and functionality. - Design Qualification (DQ) which verifies that the design will meet the requirements in the URS. - Installation Qualification (IQ) which verifies proper installation. - Operational Qualification (OQ) which tests the operation of the equipment. - Performance Qualification (PQ) which verifies the equipment can perform as intended based on approved processes and specifications. Guidance and requirements for each qualification type are defined. Supporting documents required for each are also listed.























































































