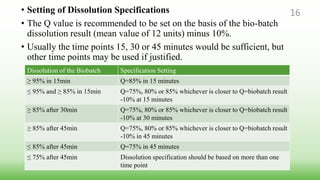
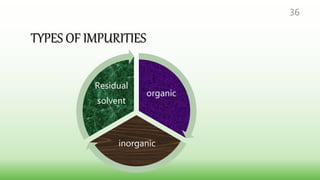
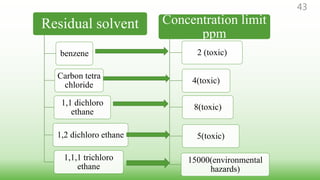
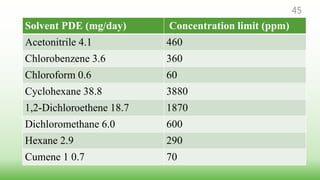
This document provides guidelines for developing specifications for new drug substances and products according to ICH Q6A. It discusses universal and specific tests/criteria that should be included for drug substances and products, such as identification, description, assay, impurities, dissolution, disintegration, content uniformity, and microbial limits. The document gives acceptance criteria and justification for key tests like dissolution, discussing how to set Q values and limits based on biobatch results and BCS classification. It also provides guidance on other tests for oral liquids, parenterals and solid dosage forms.