Downloaded 32 times
















The document discusses various computational structural biology methods for modeling and analyzing protein structures, highlighting techniques such as comparative modeling, de novo modeling, and integrative methods. It stresses the importance of template selection and alignment accuracy in comparative modeling and outlines challenges faced in structure determination. Additionally, it covers the role of structural genomics and cryoelectron microscopy in advancing our understanding of complex macromolecular assemblies.
Overview of Computational Structural Biology methods and applications by Antonio E. Serrano.
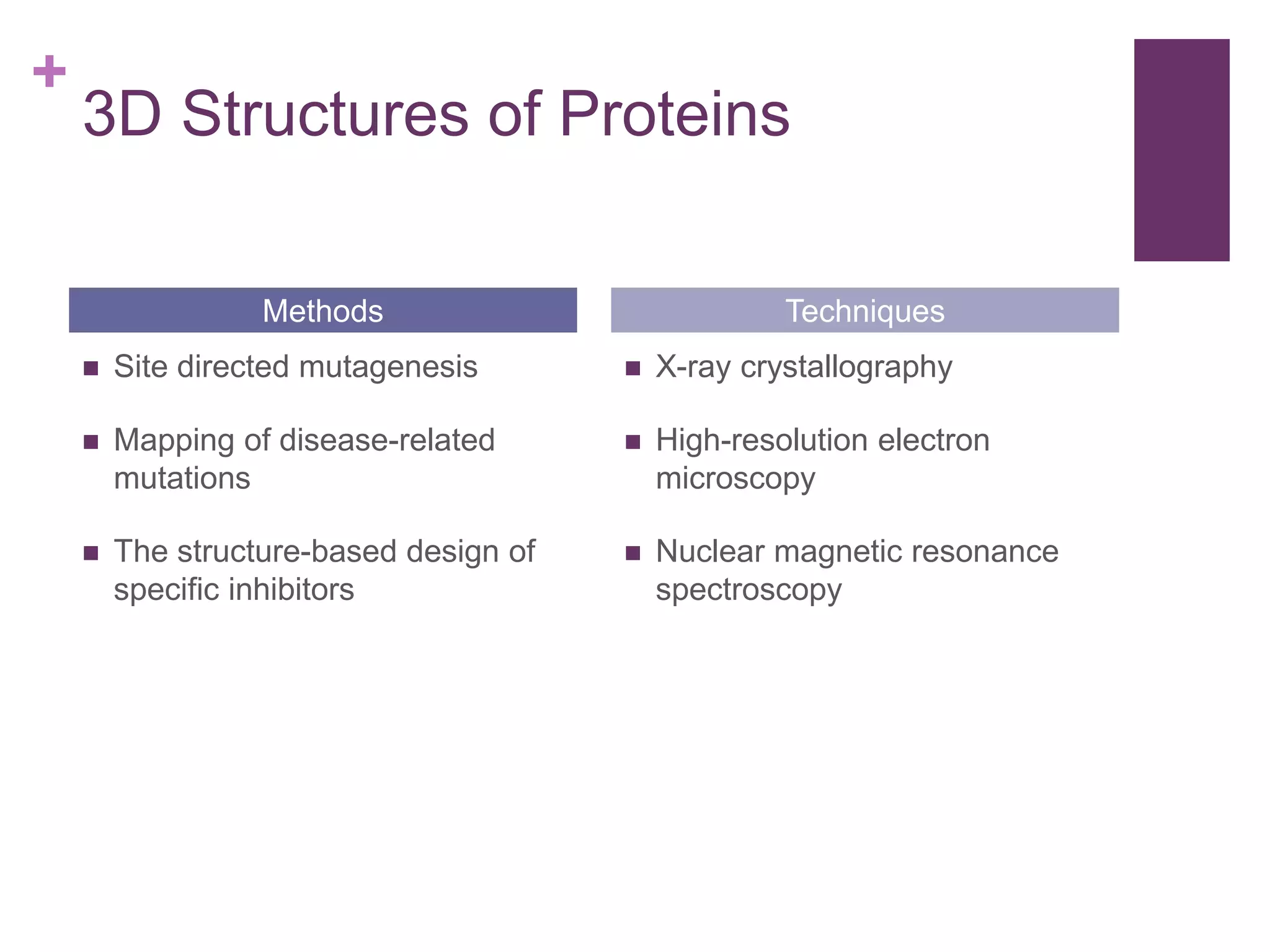
Key methods for analyzing protein 3D structures, including mutagenesis, X-ray crystallography, and spectroscopy.
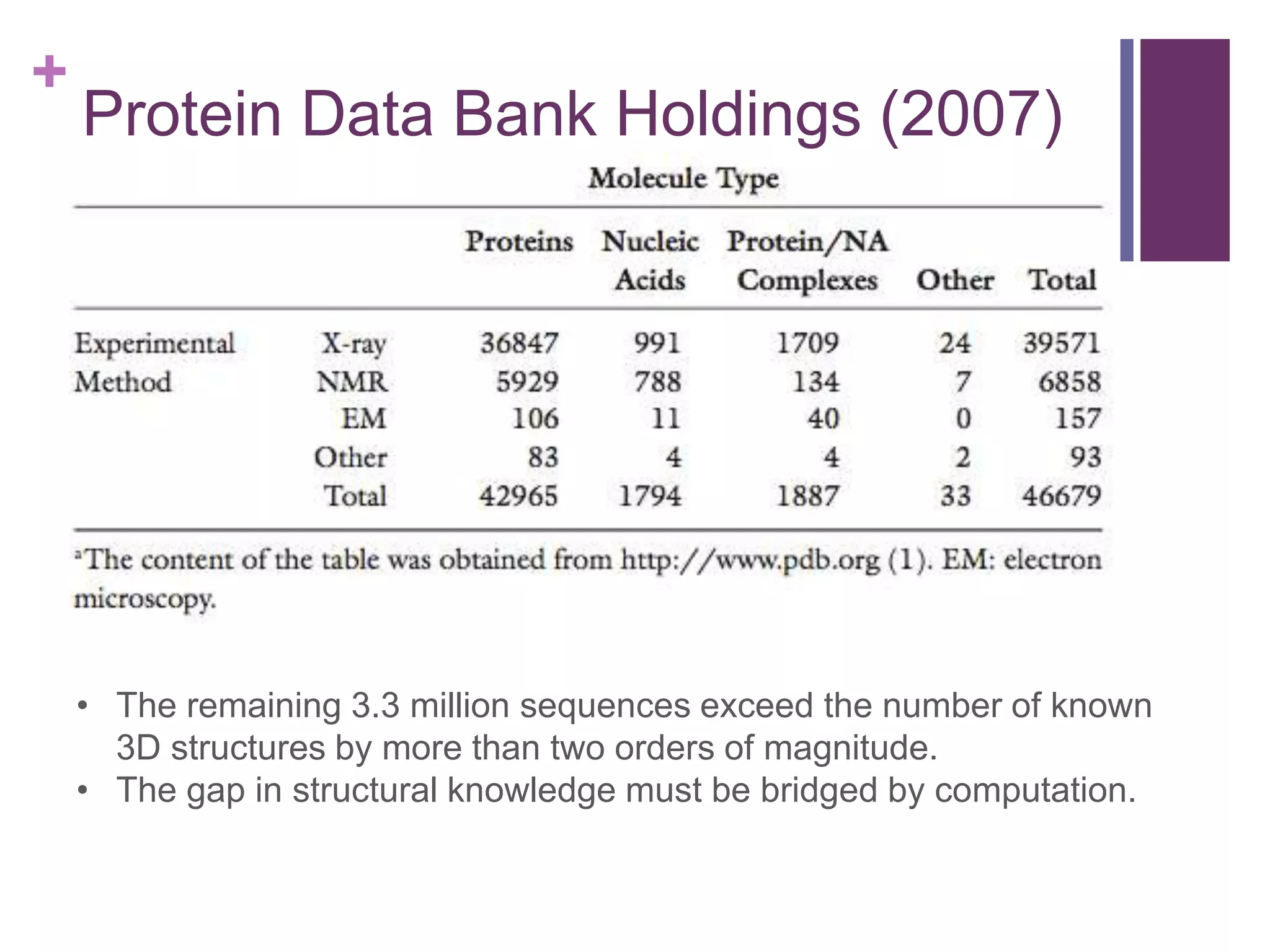
In 2007, 3.3 million sequences exceeded known 3D structures, highlighting the need for computational methods.
Different protein modeling methods: comparative, fold recognition, de novo, and integrative modeling.
Steps in comparative modeling, including template identification, alignment, modeling, refinement, evaluation.
Metrics for evaluating target-template sequence identity and its impact on model accuracy.
Common alignment techniques for protein modeling including PSI-Blast, HMM, and profile-based methods.
Two main approaches for modeling conserved regions in protein structures and optimization strategies.
Methods for refining protein models and assessing their accuracy against native structures.
Limitations and accuracy factors in comparative modeling dependent on sequence identity levels.
De novo modeling does not use known structures as templates; aims to predict protein structure from sequence.
Rosetta method's application in de novo prediction with high accuracy but significant computational demands.
Significance of three-dimensional atomic-level structure determination in comparative modeling.
Efforts in structural genomics to determine protein structures efficiently; accounts for 20% of new structures.
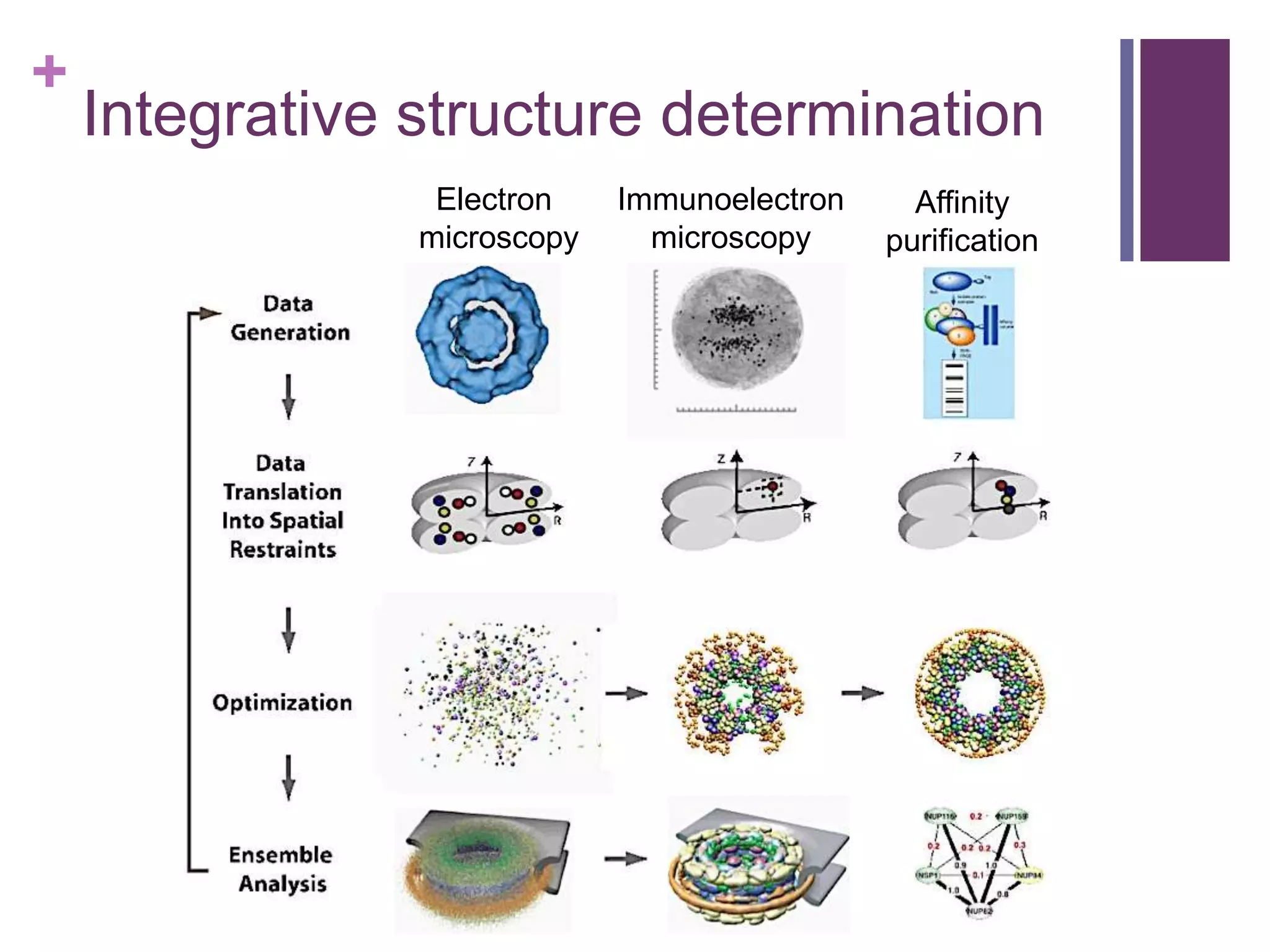
Hybrid approaches for characterizing biological interactions, enhancing traditional structural biology methods.
Techniques like electron microscopy and affinity purification for detailed structural determination.
Uses of protein modeling in mutagenesis, engineering, drug discovery, and studying mutations.
Overview of available protein modeling servers and software tools for structural biology.
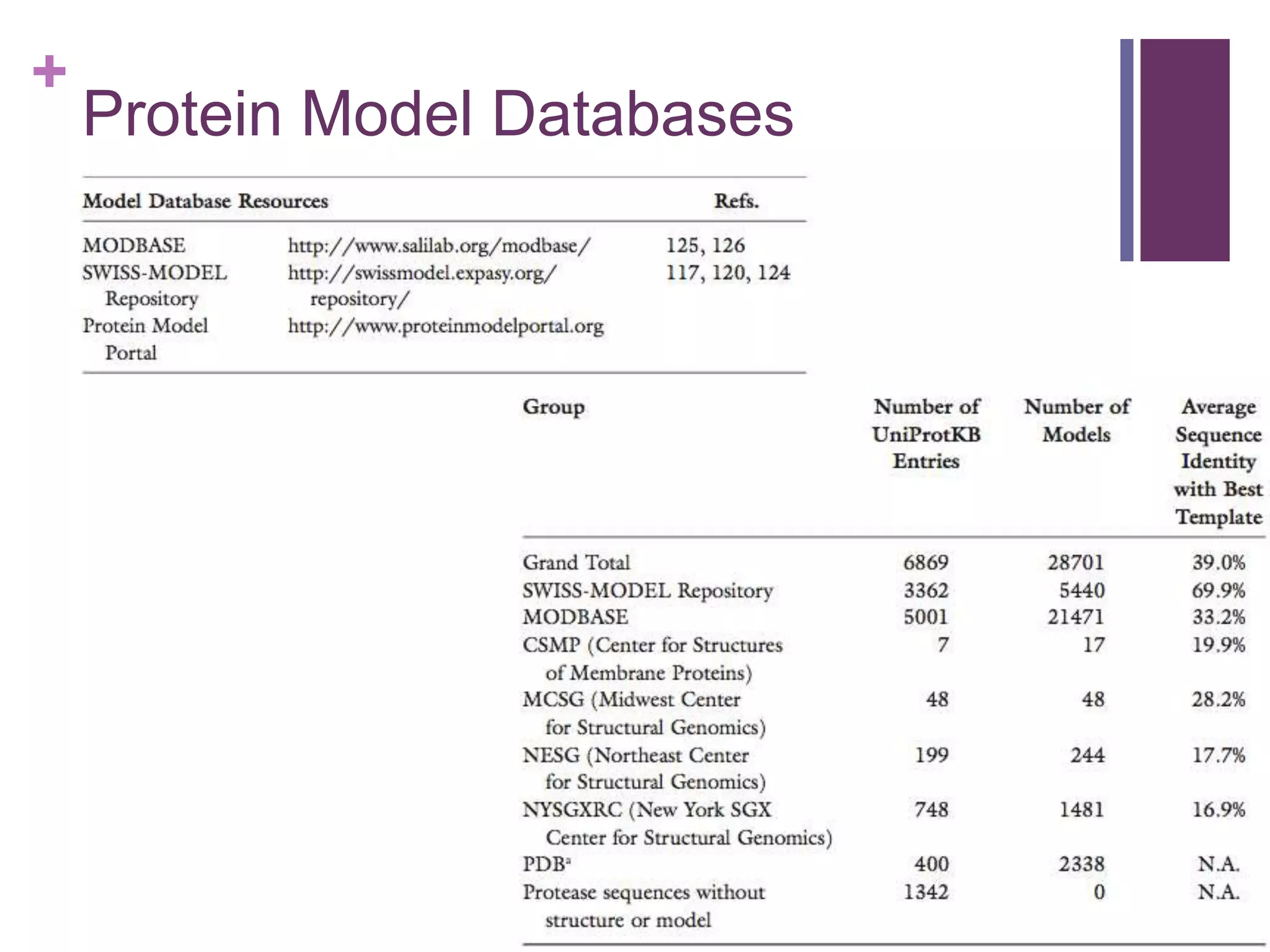
Discussion on databases available for storing and sharing protein models and structures.
Cryoelectron microscopy's role in studying large, complex macromolecules and various biological structures.











































































































