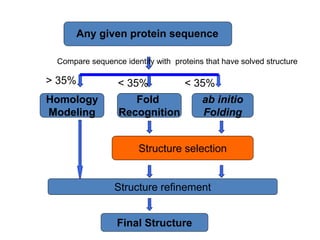
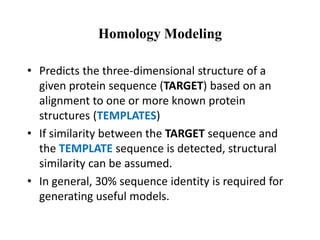
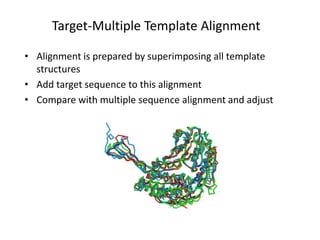
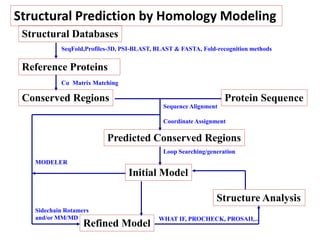
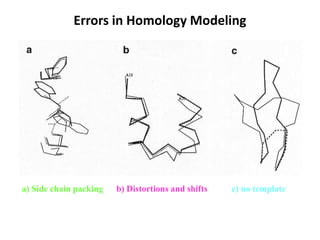
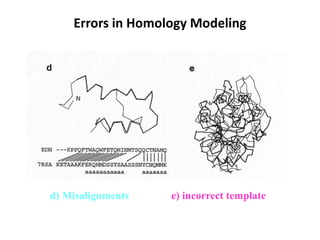
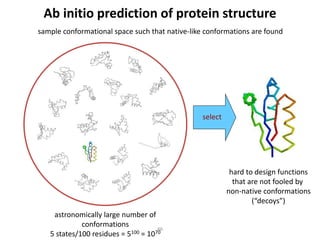
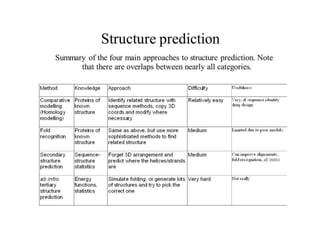
The document discusses protein structure modeling through homology modeling. It describes the key steps in homology modeling which include: (1) finding a suitable template through database searches, (2) aligning the target sequence to the template, (3) assigning coordinates from conserved regions of the template, (4) building loops and variable regions either from other structures or de novo, (5) searching for optimal side chain conformations, and (6) refining the model through molecular mechanics. The document emphasizes validating the final model to identify any inherent errors from the template or modeling process.




























































































![Polymer [ बहुलक ] Chemistry Notes PDF - Irfanullah Mehar - JJ Sir Chemistry.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/polymerchemistrynotespdf-irfanullahmehar-jjsirchemistry-260210172118-3f9b37f7-thumbnail.jpg?width=640&height=640&fit=bounds)









