Downloaded 11 times













![Protein Folding
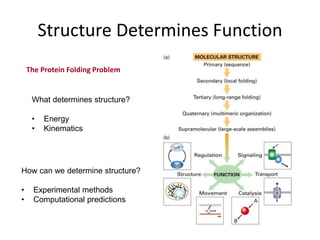
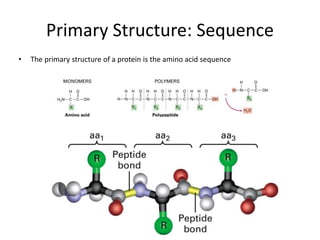
• The amino-acid sequence of a protein determines the 3D fold [Anfinsen et
al., 1950s]
Some exceptions:
– All proteins can be denatured
– Some proteins have multiple conformations
– Some proteins get folding help from chaperones
• The function of a protein is determined by its 3D fold
• Can we predict 3D fold of a protein given its amino-acid sequence?](https://image.slidesharecdn.com/structuralpredection-211120125054/85/Protein-Structural-predection-16-320.jpg)
























The document provides an overview of protein structural prediction, emphasizing the hierarchical nature of protein structure, which is determined by the amino acid sequence and various interactions. It discusses methodologies for determining protein structure, including experimental techniques like X-ray crystallography and computational approaches such as homology modeling and ab initio prediction. Additionally, it highlights the challenges of protein folding, the importance of energy interactions, and the role of databases in classifying protein structures.



































































