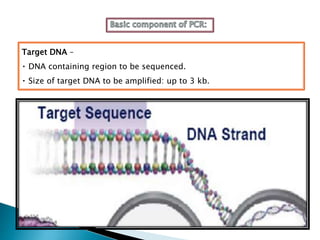
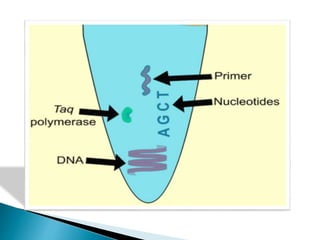
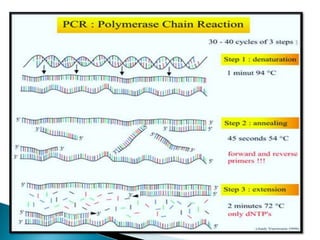
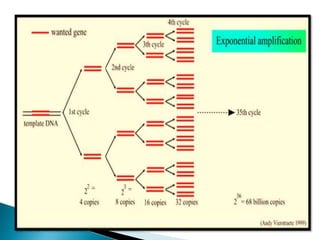
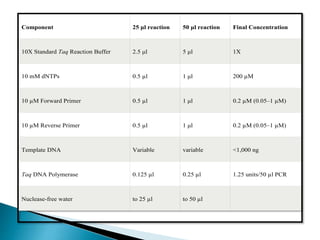
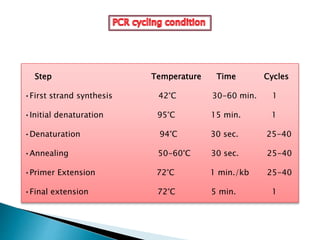
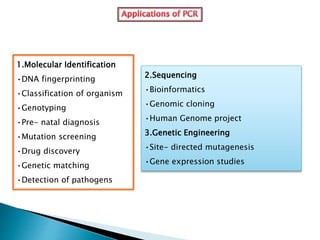
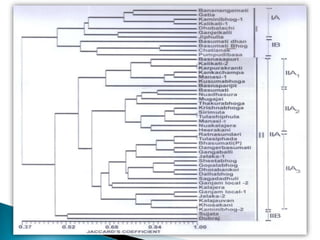
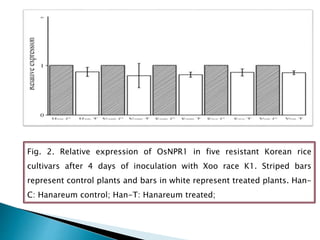
The document summarizes a seminar presentation on PCR and its different types. It begins by defining PCR and its history. It then discusses the basic components, procedures/steps, principles, and instrumentation of PCR. It describes different types of PCR including standard PCR, nested PCR, real-time PCR, and reverse transcriptase PCR. It outlines several applications of PCR and concludes by discussing the advantages and disadvantages of PCR.