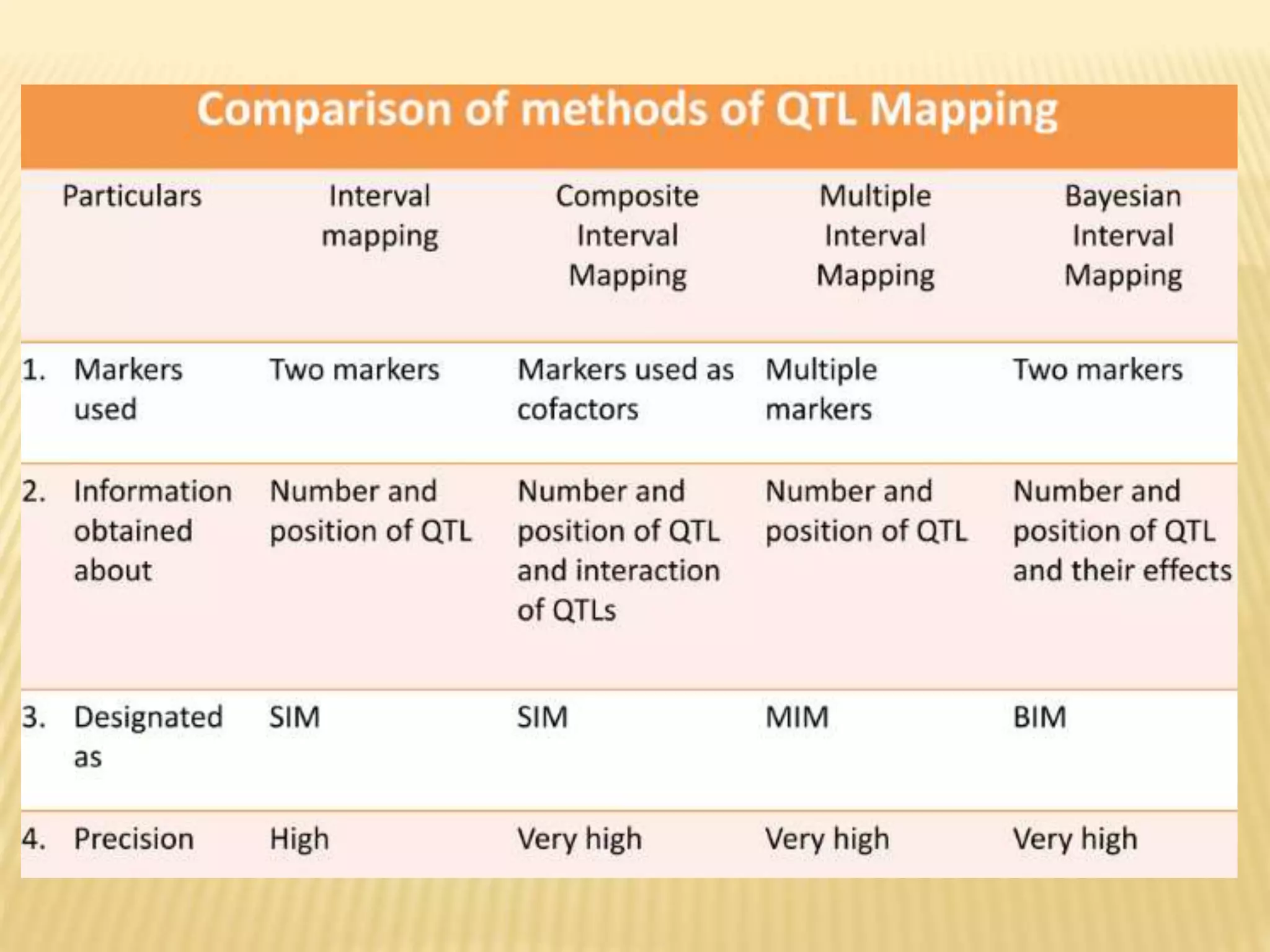
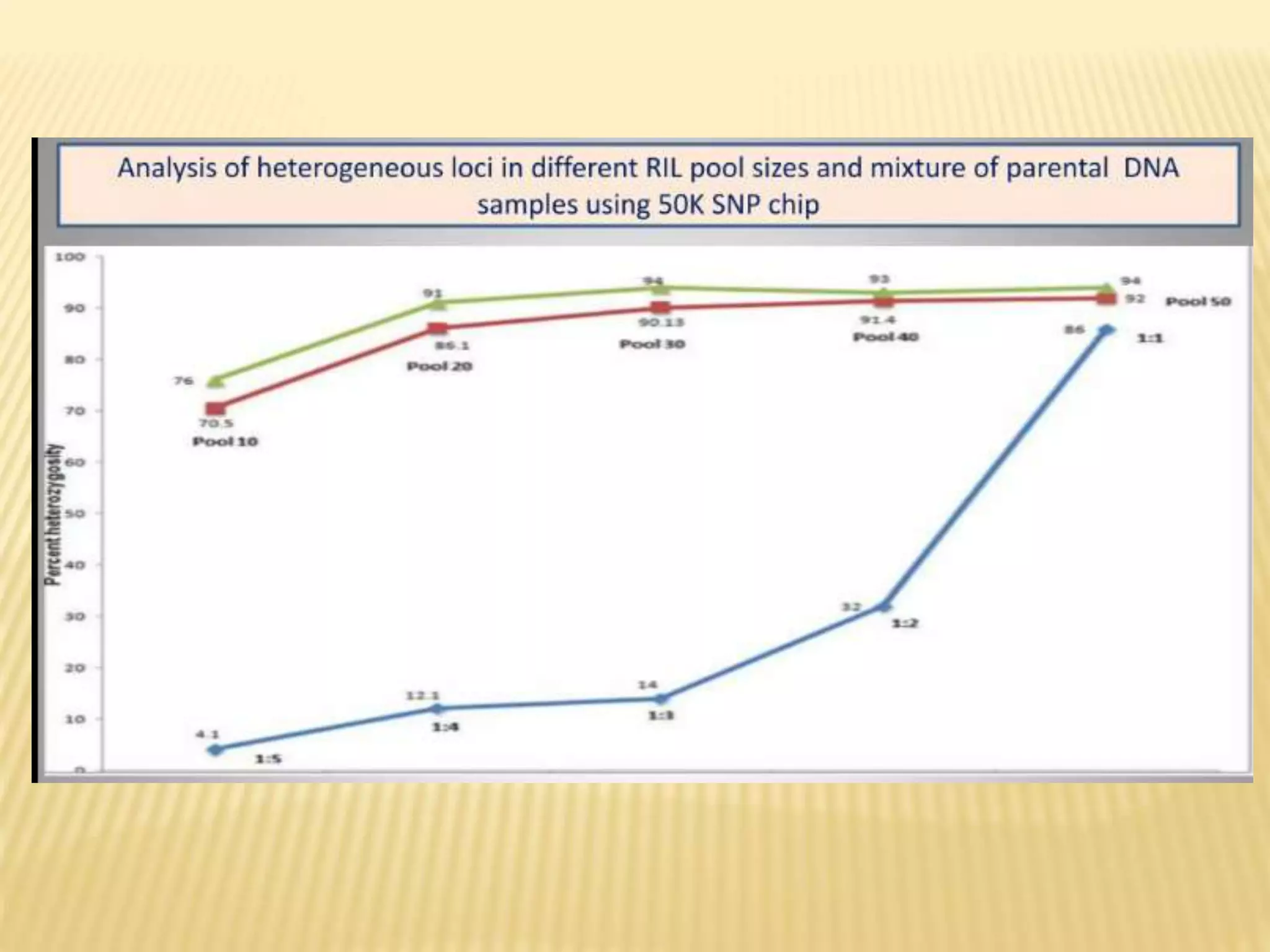
This document discusses quantitative trait loci (QTL) mapping. It begins by defining QTLs as genomic regions associated with quantitative traits. The document then covers the principles, objectives, methods, and steps of QTL mapping. It discusses techniques like single marker analysis, interval mapping, composite interval mapping, and Bayesian interval mapping. The document also presents a case study on QTL mapping for salt tolerance in rice using bulked segregant analysis of recombinant inbred lines. It finds 34 QTL regions, confirming previously identified regions and identifying new QTLs.