Downloaded 29 times


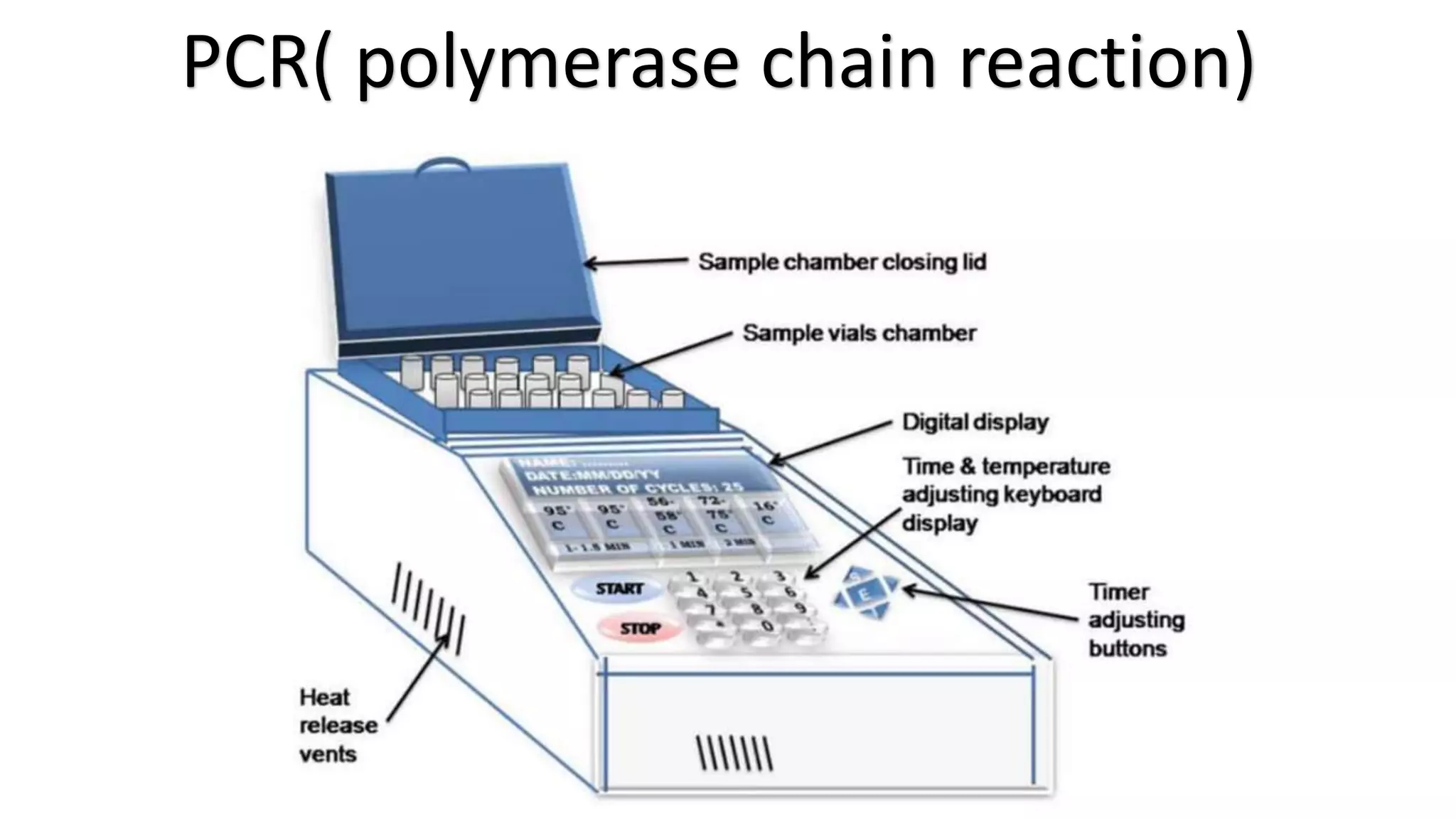
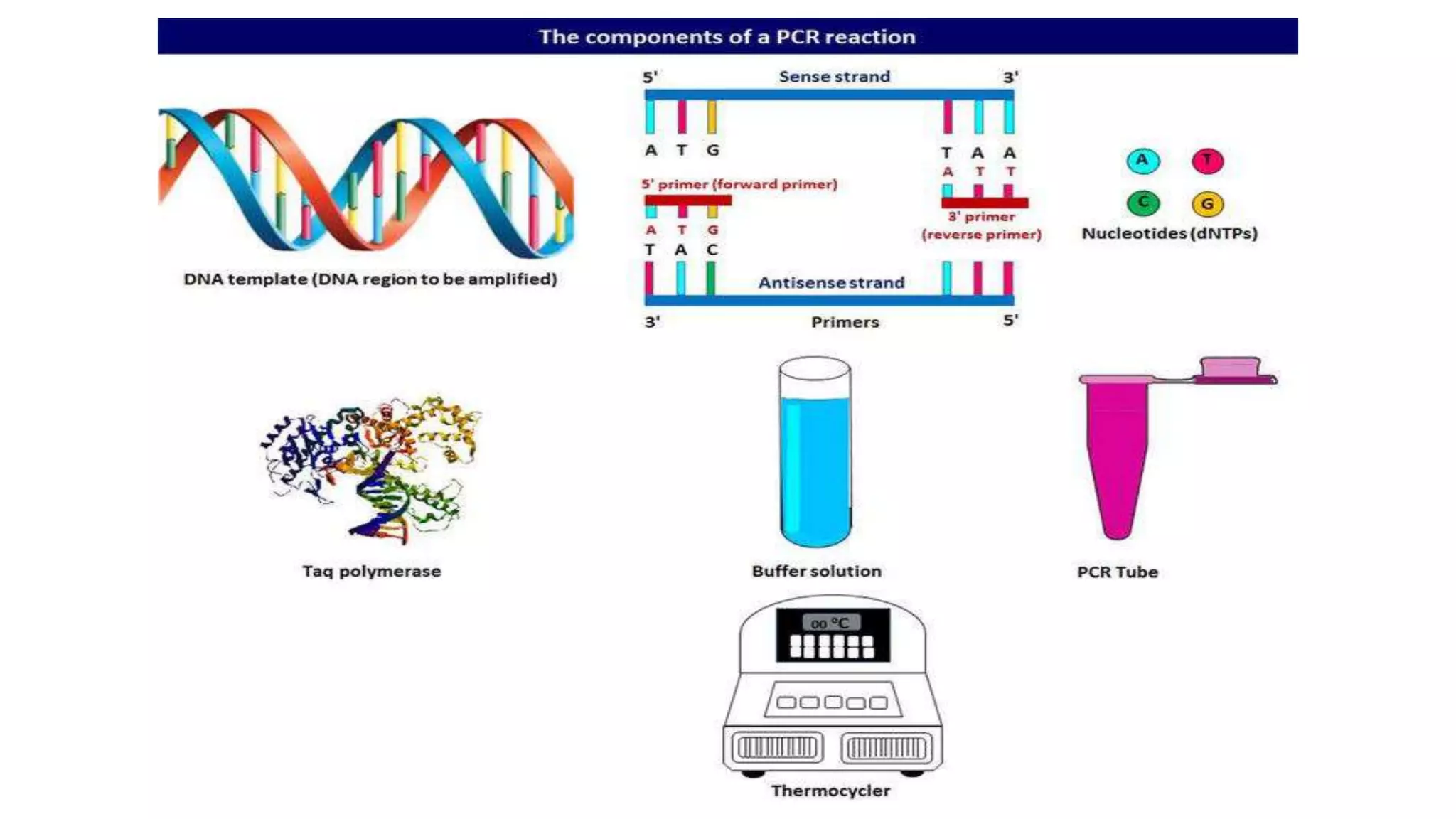

















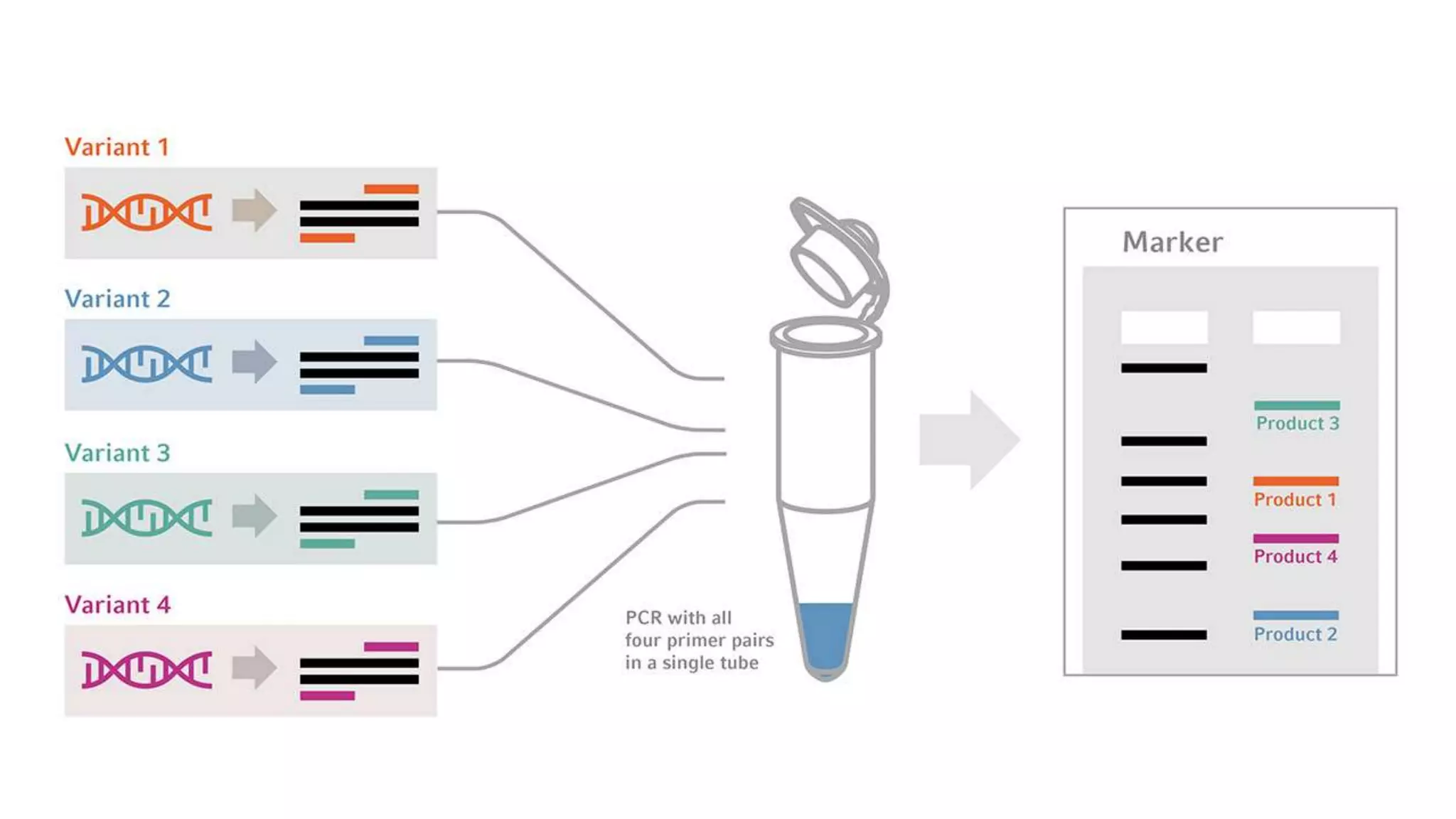






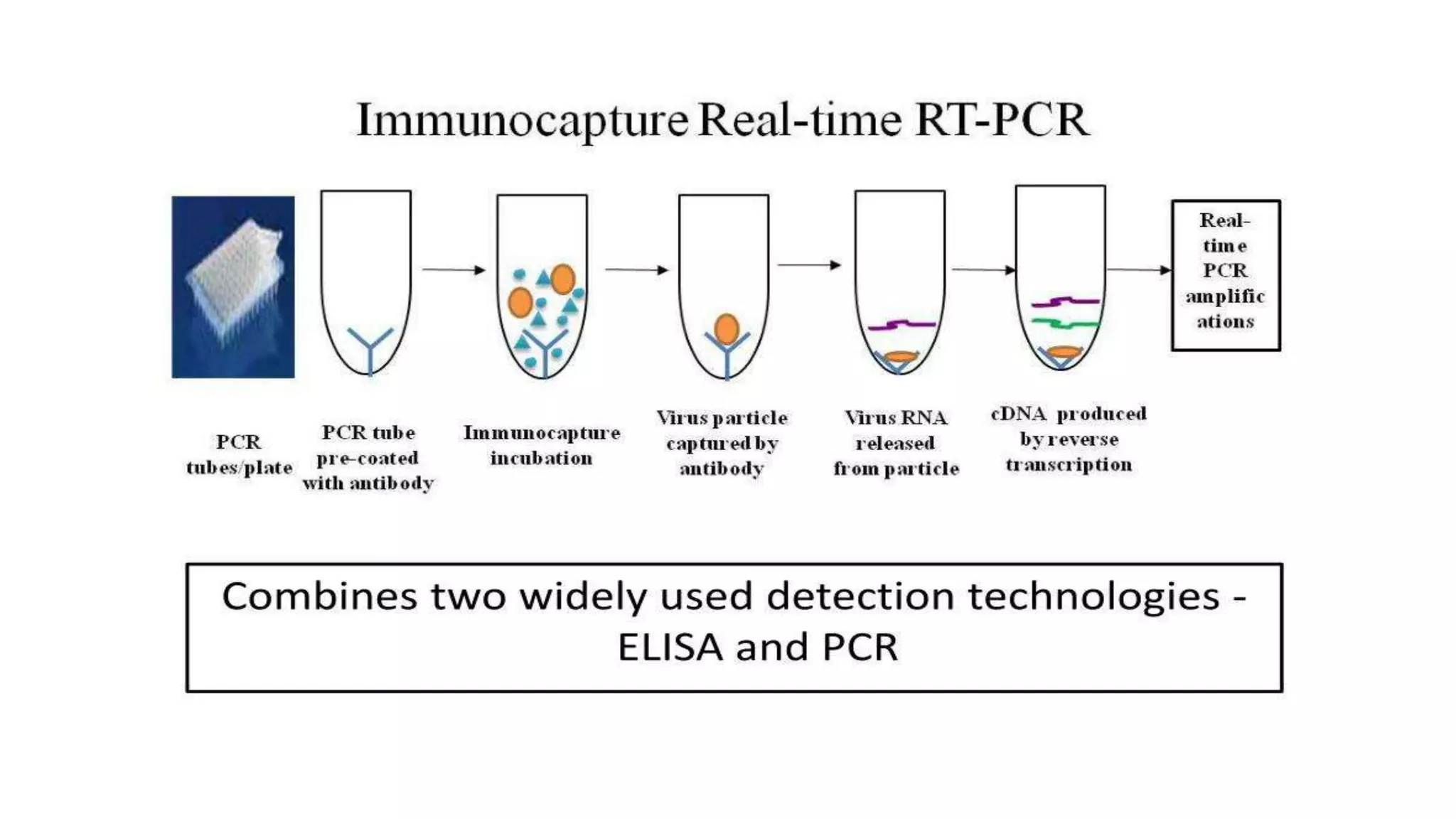












The document provides an overview of Polymerase Chain Reaction (PCR), a critical molecular technique developed by Kary Mullis in 1983 for amplifying DNA, with applications in medical and biological research, such as disease diagnosis and DNA sequencing. It discusses key components including DNA template, polymerases, primers, and the three main steps of the PCR process: denaturation, annealing, and extension, along with various types of PCR used in plant pathology. Additionally, it details procedures for extracting DNA from fungal pathogens and plant tissues, and methods for amplifying specific DNA regions.


















































































