Downloaded 52 times



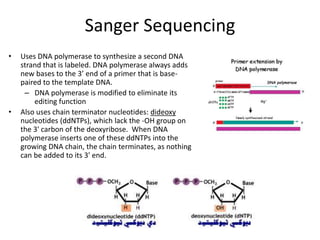

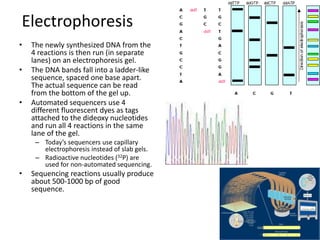
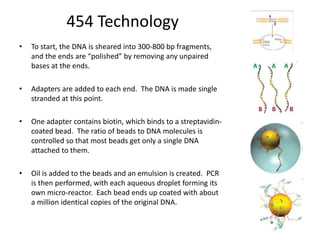
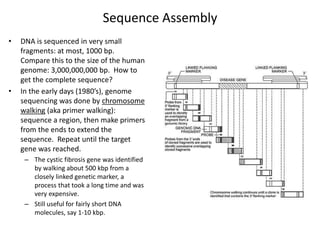


This document discusses several methods for gene sequencing, including Sanger sequencing, 454 technology, and sequence assembly. Sanger sequencing uses DNA polymerase and chain terminating nucleotides to synthesize labeled DNA strands of varying lengths that can then be separated by electrophoresis to determine the sequence. 454 technology attaches DNA fragments to beads and performs emulsion PCR to generate millions of copies for pyrosequencing. Sequence assembly involves combining overlapping sequences from fragmented DNA reads to reconstruct the full genome sequence, as individual reads are usually only 500-1000 base pairs long.























































































