











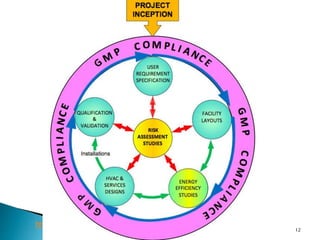
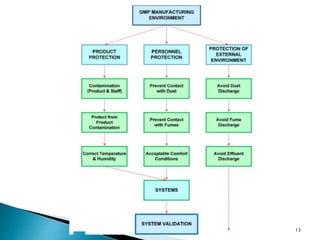










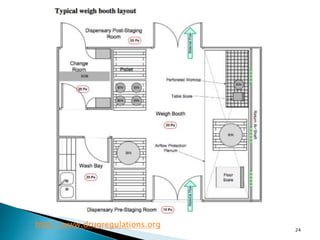
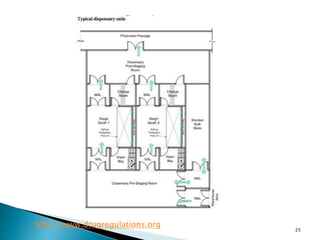

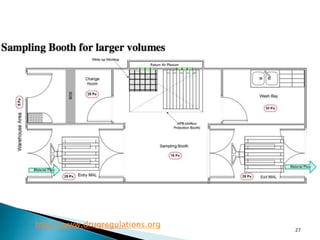
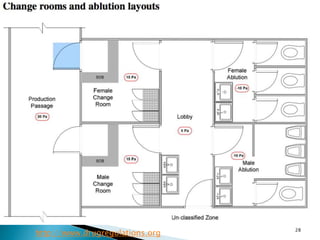
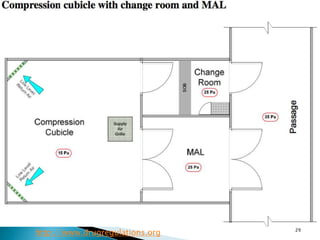
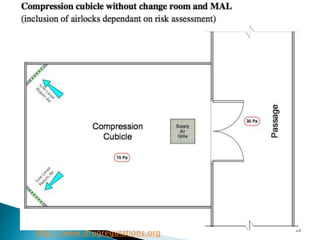
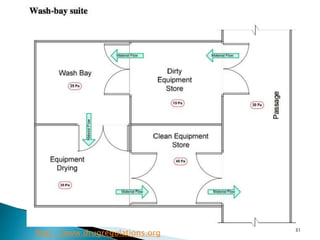









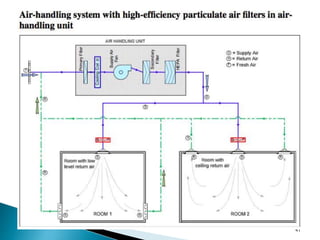

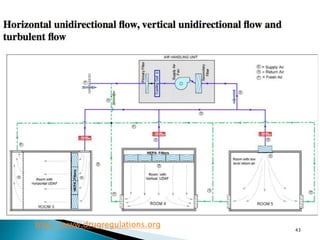





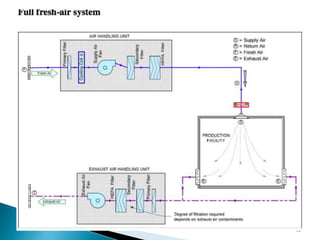
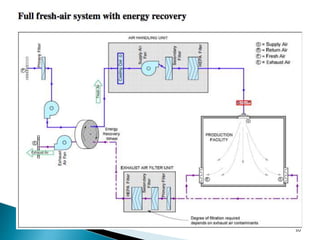


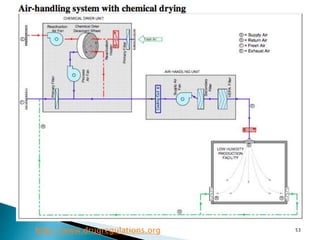













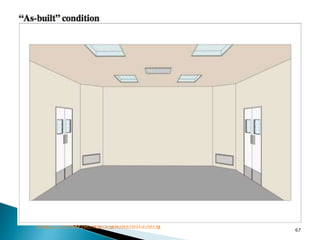
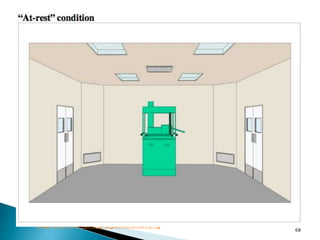

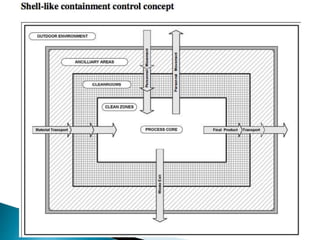

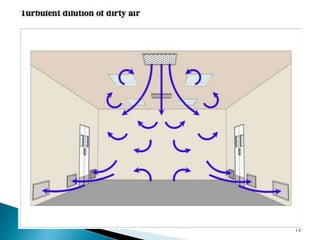
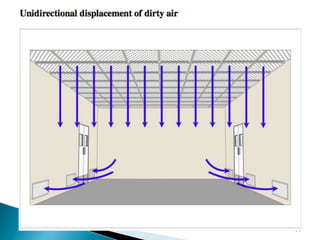

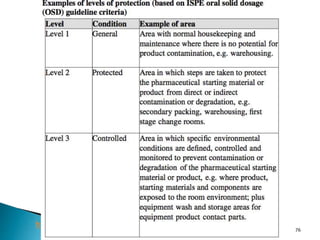




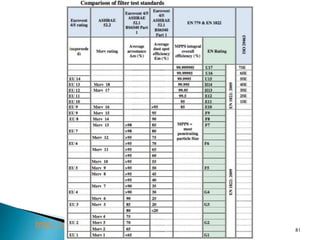
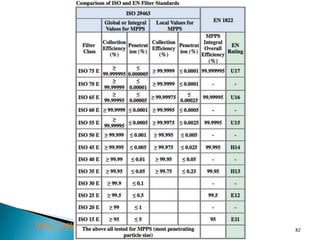

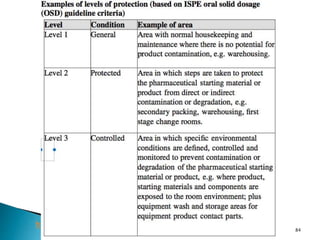



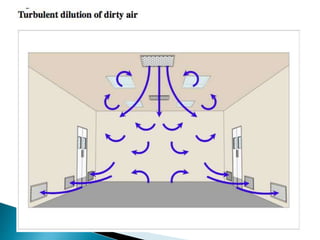
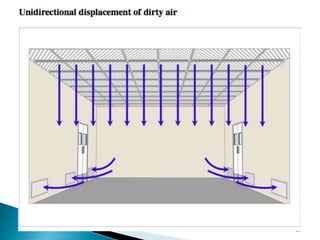


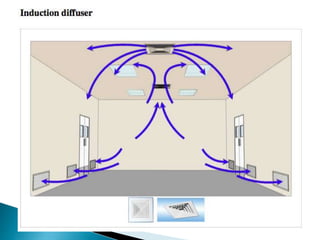
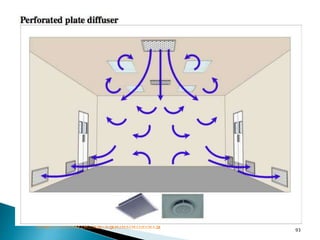
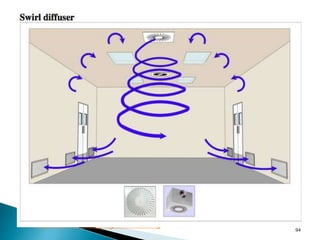




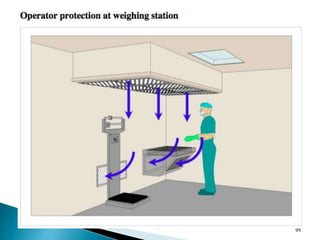




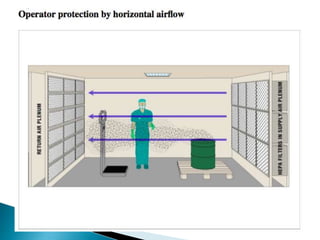


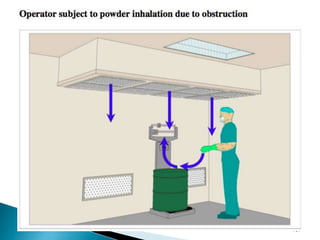

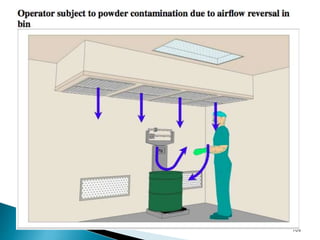
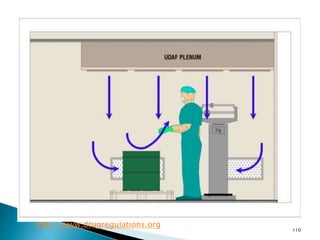

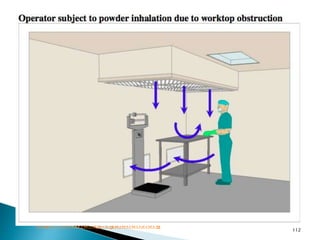

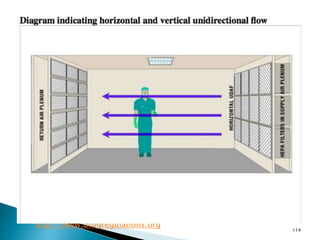
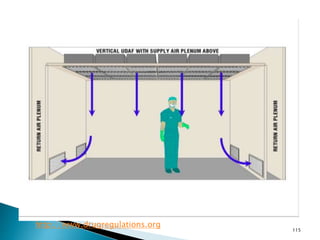














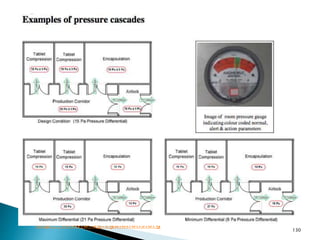



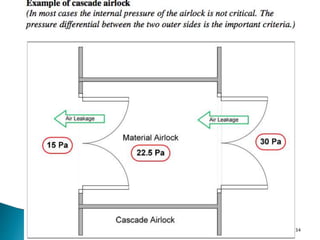
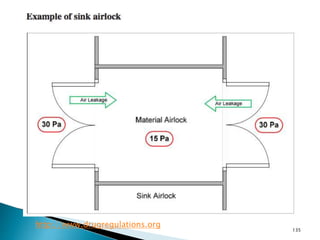
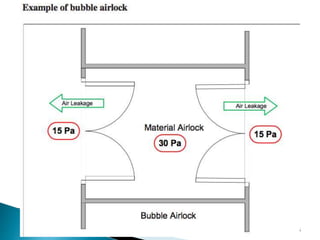


































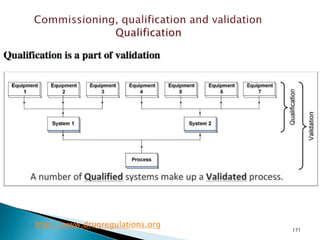

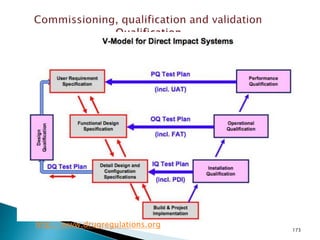










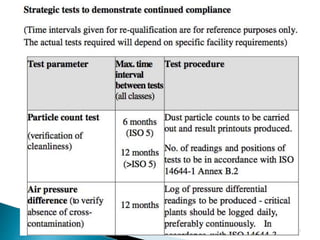


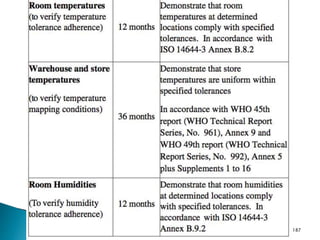



















This document summarizes guidelines from the World Health Organization (WHO) regarding heating, ventilation and air conditioning (HVAC) systems for manufacturing solid dosage form pharmaceuticals. It discusses how HVAC design influences architectural layouts and contamination control. Key factors in HVAC system design include temperature and humidity control, airflow patterns, and filtration to maintain air cleanliness without compromising product quality or safety.

















































































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)





