Downloaded 1,039 times



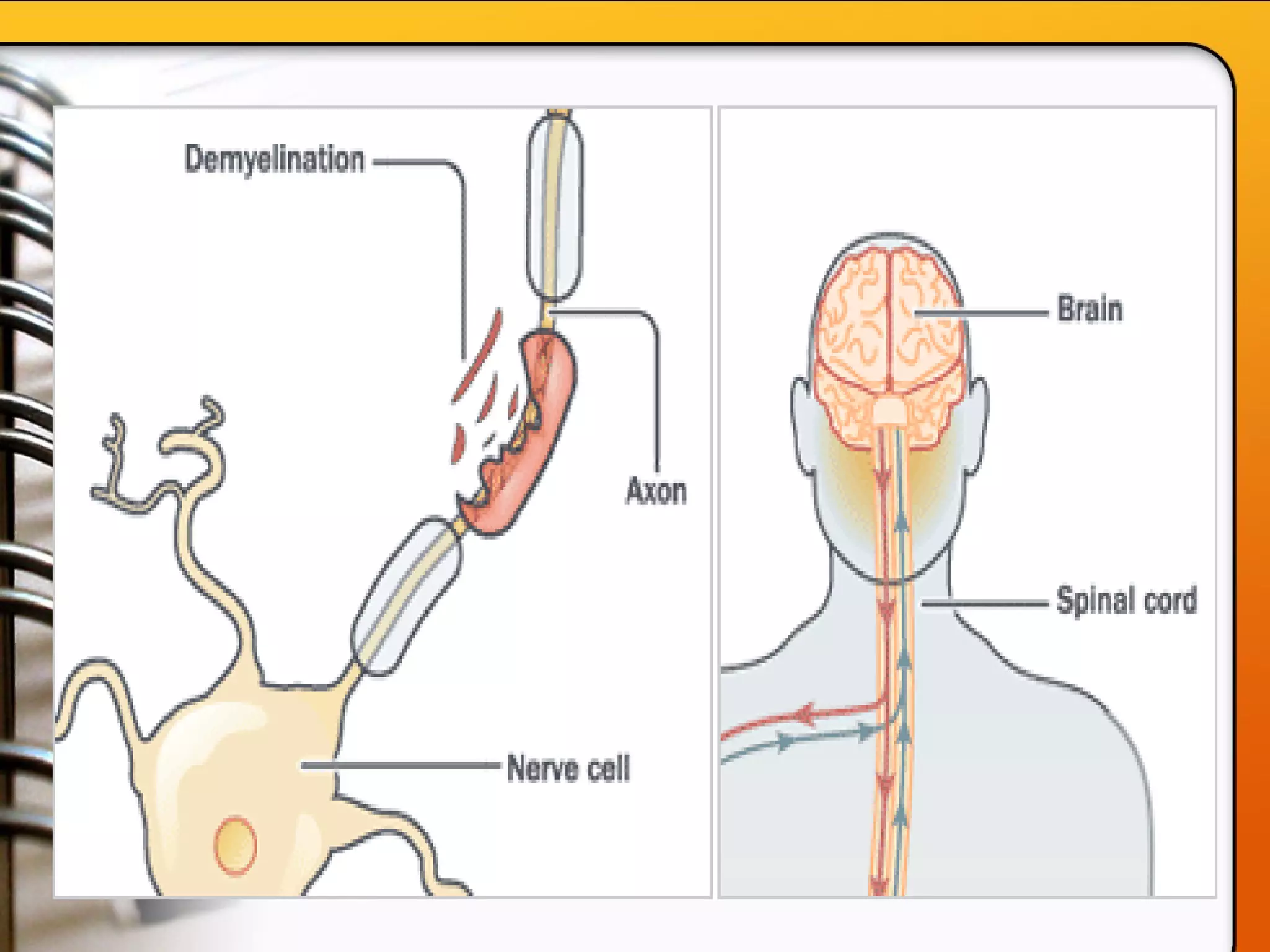













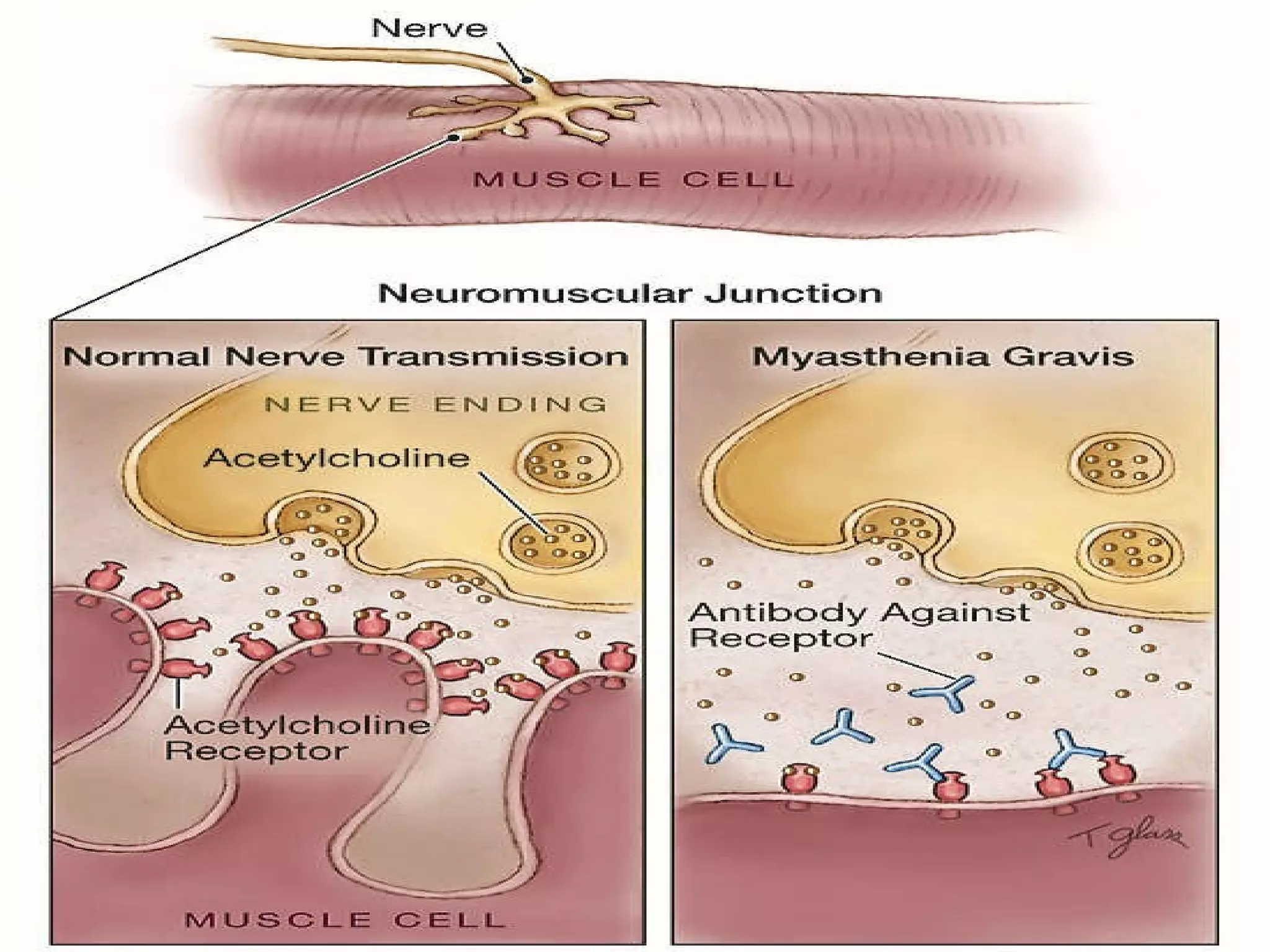




























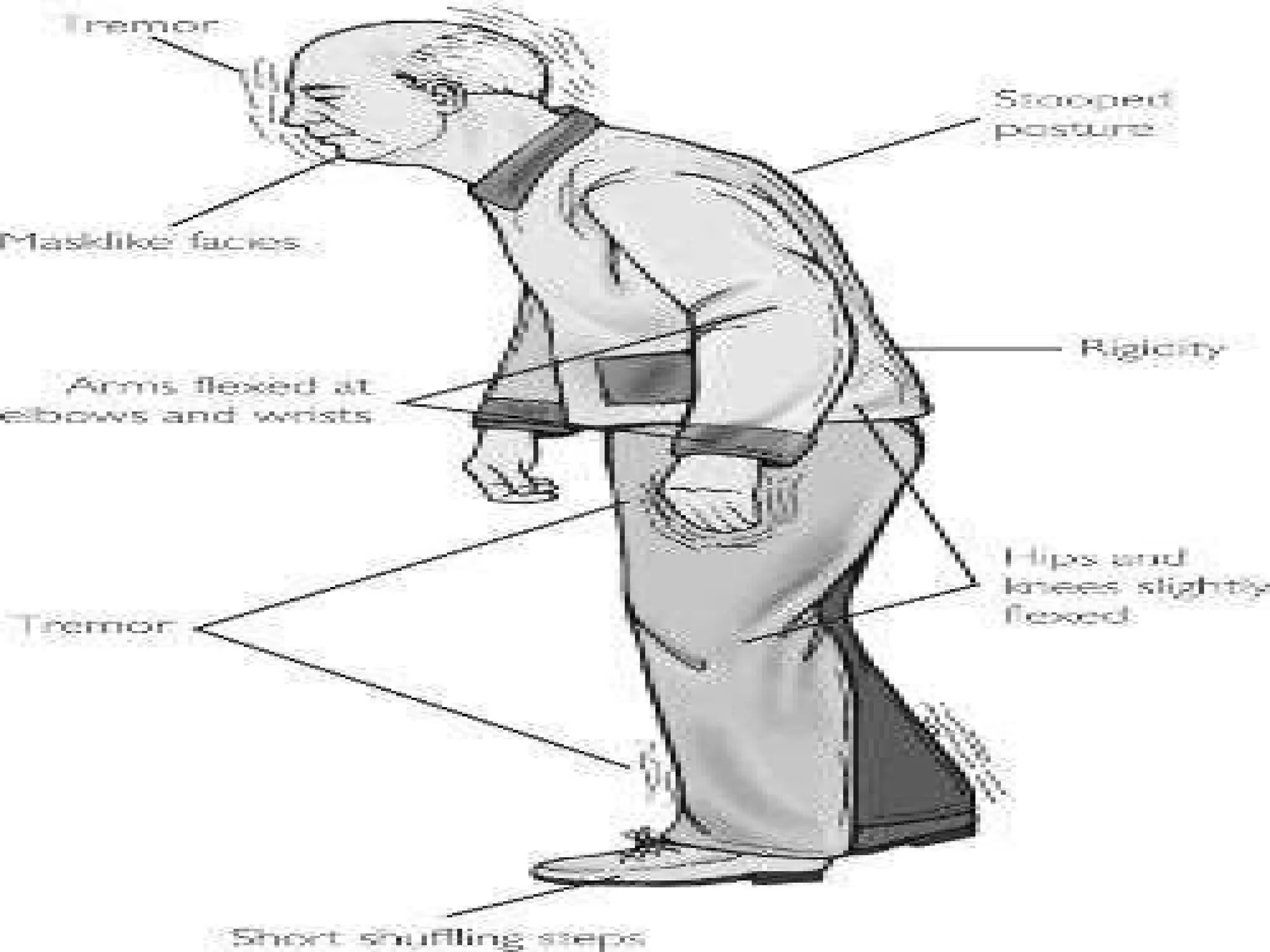





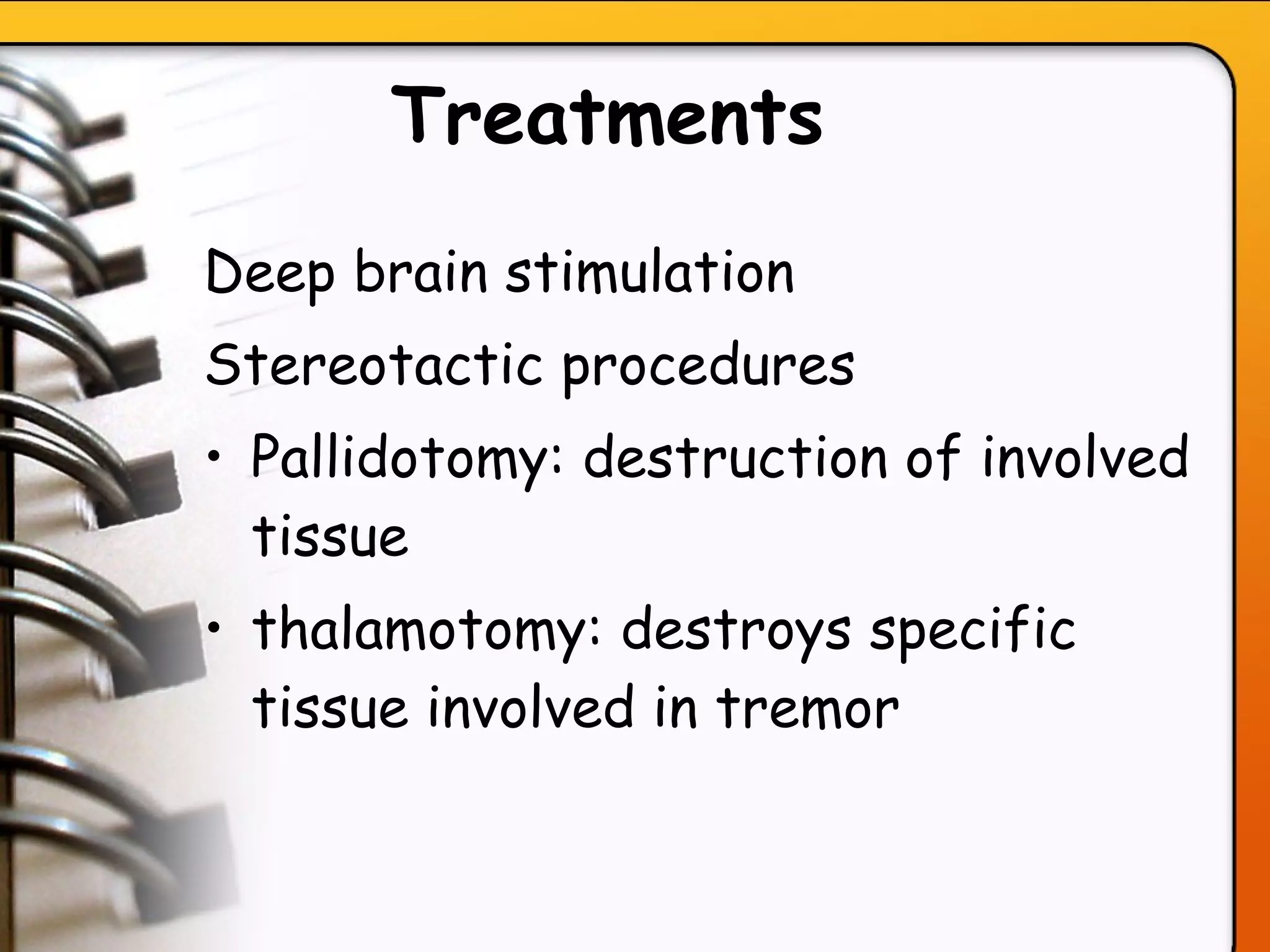










































This document provides an overview of several neurologic disorders, including: - Multiple sclerosis, which causes fatigue, vision issues, weakness and more. Treatment focuses on retaining function and limiting disability. - Myasthenia gravis, an autoimmune disorder causing severe muscle weakness. Medications aim to improve symptoms. - Guillain-Barré syndrome, an acute inflammatory disorder causing ascending paralysis. Supportive care and monitoring of respiratory function are priorities. - Parkinson's disease, characterized by tremors and rigidity. Medications may provide relief but symptoms gradually worsen over time. - Huntington's disease, an inherited disorder causing chorea and dementia. No cure exists, and



































![Trends and issues in nursing [compatibility mode]](https://cdn.slidesharecdn.com/ss_thumbnails/trendsandissuesinnursingcompatibilitymode-120712052718-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)




































































