


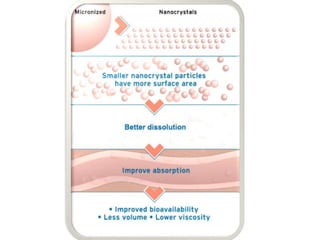


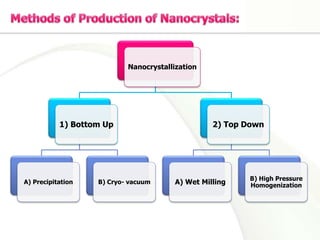





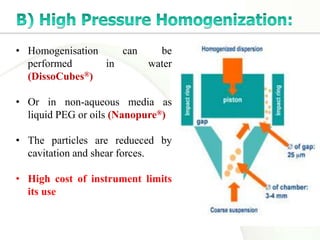







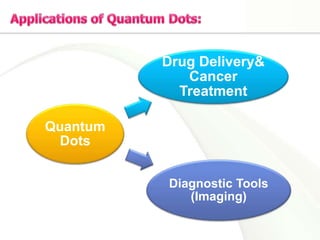
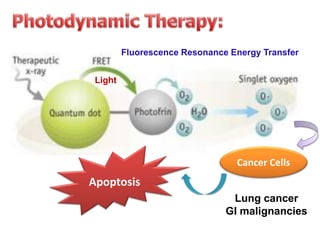

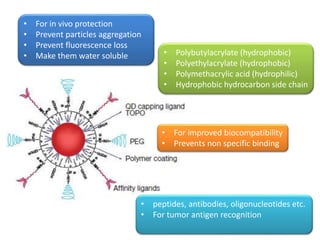
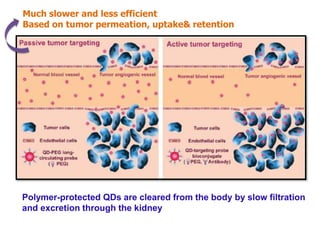



Nanocrystals are pure drug particles in the nanometer size range that can increase drug solubility and bioavailability without using surfactants. Various "bottom up" and "top down" methods are used to produce drug nanocrystals including precipitation, cryo-vacuum processing, wet milling, and high pressure homogenization. Drug nanocrystals have potential applications for oral, transdermal, and targeted cancer delivery and imaging. Further research is still needed to reduce nanocrystal toxicity before clinical use.





























































































