Downloaded 251 times





![6
Test method
• A definitive procedure for the identification, measurement, and evaluation of a material,
product, system, or service that produces a test result. [ASTM D4392-87]
• The appropriate methods should include:-
o Sampling,
o Handling,
o Transport,
o Storage and
o Sample preparation.
The laboratory shall have instructions on the use and operation of all relevant equipment.[ISO 17025]](https://image.slidesharecdn.com/methoddevelopment-170721132459/85/Method-development-6-320.jpg)
![7
Minimum method information
a) Appropriate identification;
b) Scope;
c) Description of the type of item to be tested or calibrated;
d) Parameters or quantities and ranges to be determined;
e) Apparatus and equipment, including technical performance requirements;
f) Reference standards and reference materials required;
g) Environmental conditions required and any stabilization period needed;
h) Description of the procedure, including
• Affixing of identification marks, handling, transporting, storing and preparation of items,
• Checks to be made before the work is started,
• Checks that the equipment is working properly and, where required, calibration and
adjustment of the equipment before each use,
• The method of recording the observations and results,
• Any safety measures to be observed;
i) Criteria and/or requirements for approval/rejection;
j) Data to be recorded and method of analysis and presentation;
k) The uncertainty or the procedure for estimating uncertainty. [ISO 17025]](https://image.slidesharecdn.com/methoddevelopment-170721132459/85/Method-development-7-320.jpg)














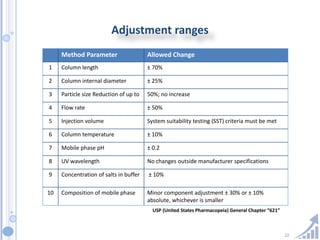









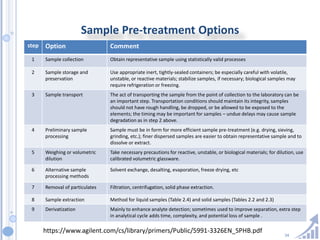





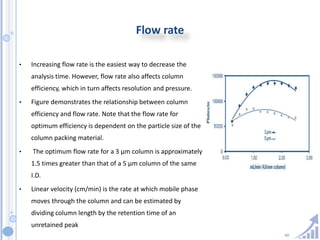


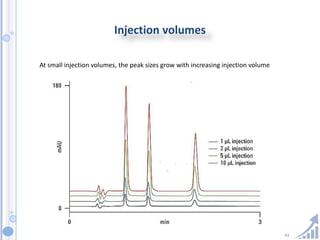
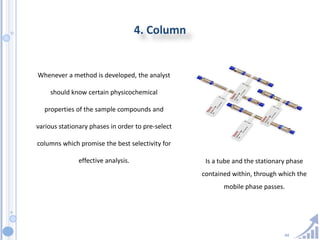
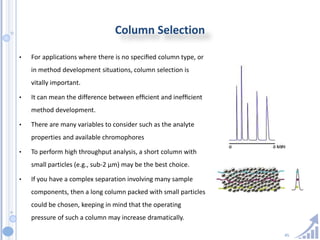





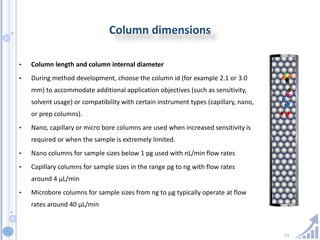

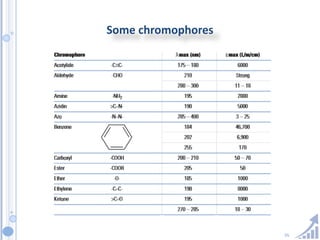

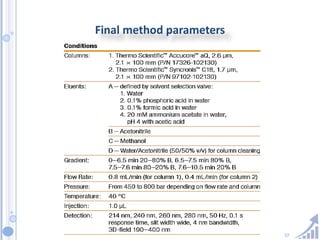

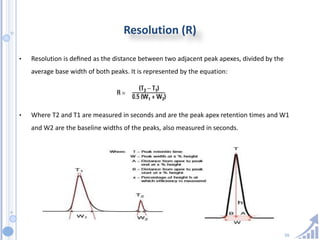


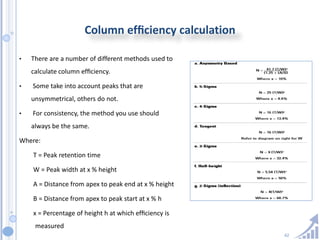
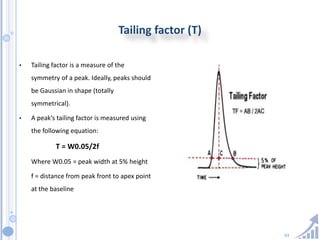
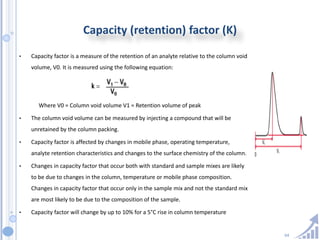


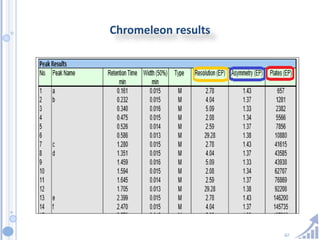
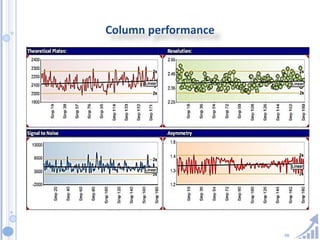



The document provides details on method development for chromatography. It discusses defining key terms, developing a test method plan, optimizing methods through experimental design techniques like factorial design. The method development process involves studying samples, setting goals, reviewing literature, selecting an approach, optimizing parameters, and finalizing the method. Critical parameters like column length and temperature, flow rate, mobile phase composition are identified for optimization. Formal validation is required once the method is developed.























































































