


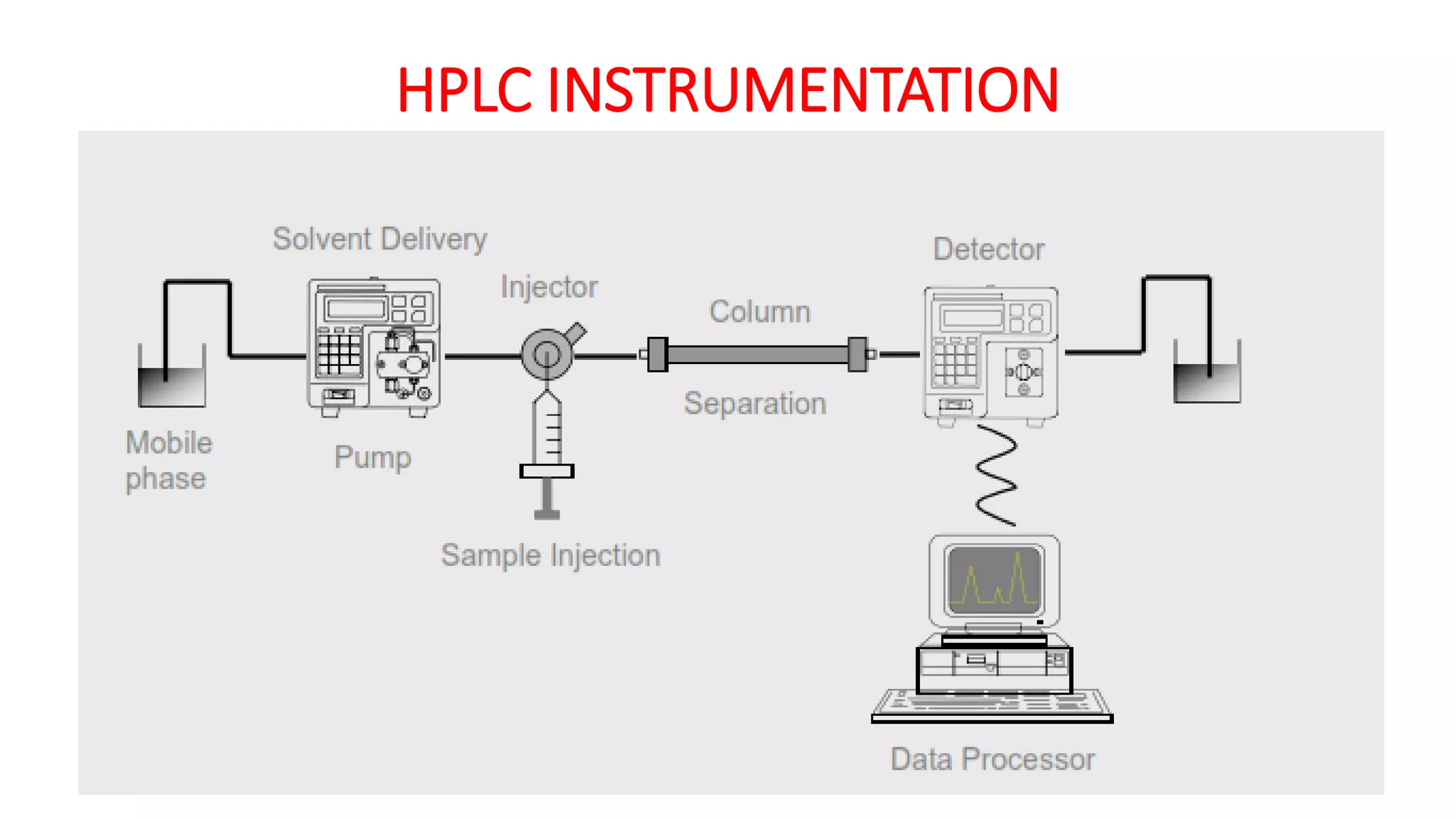
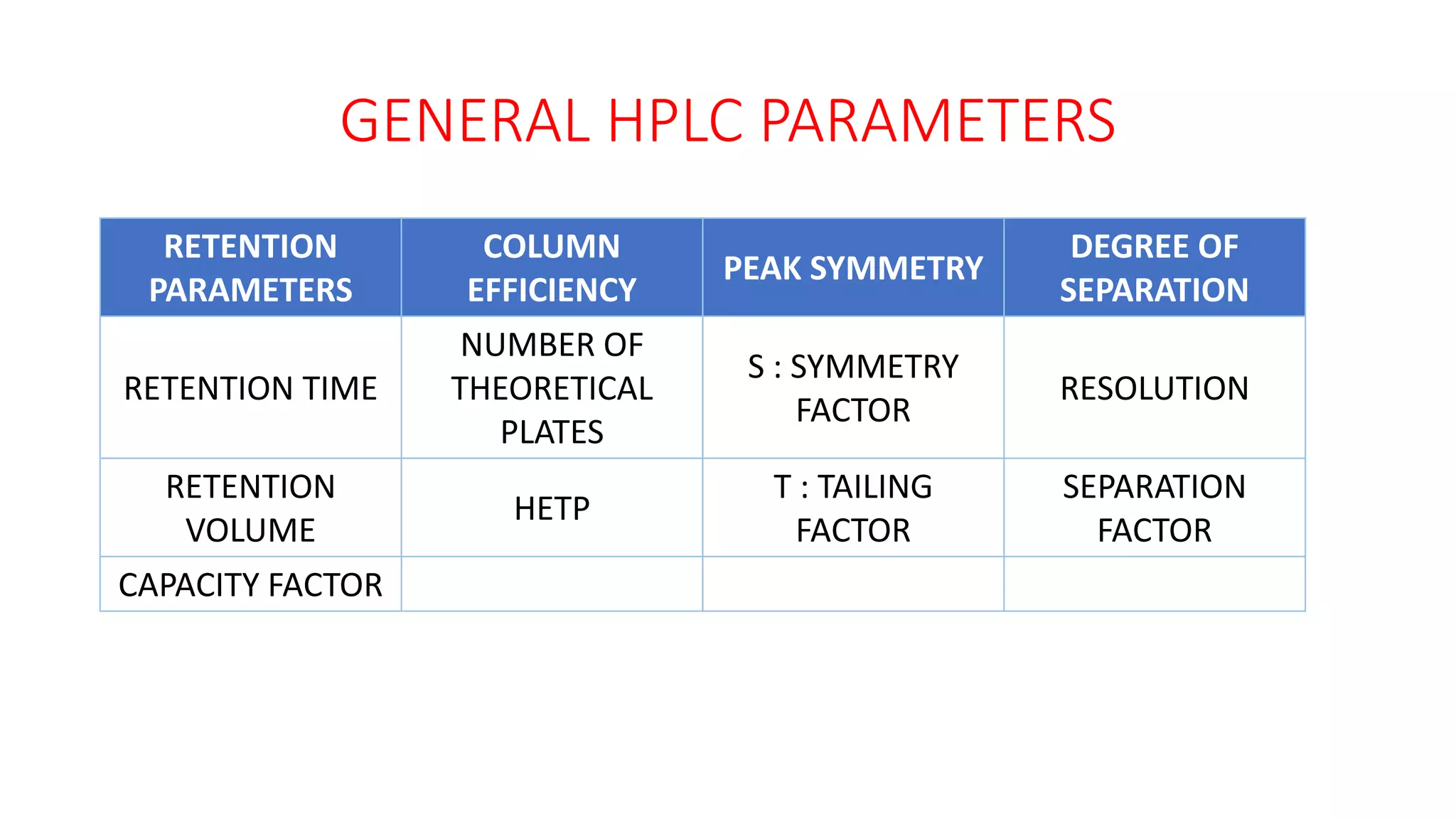

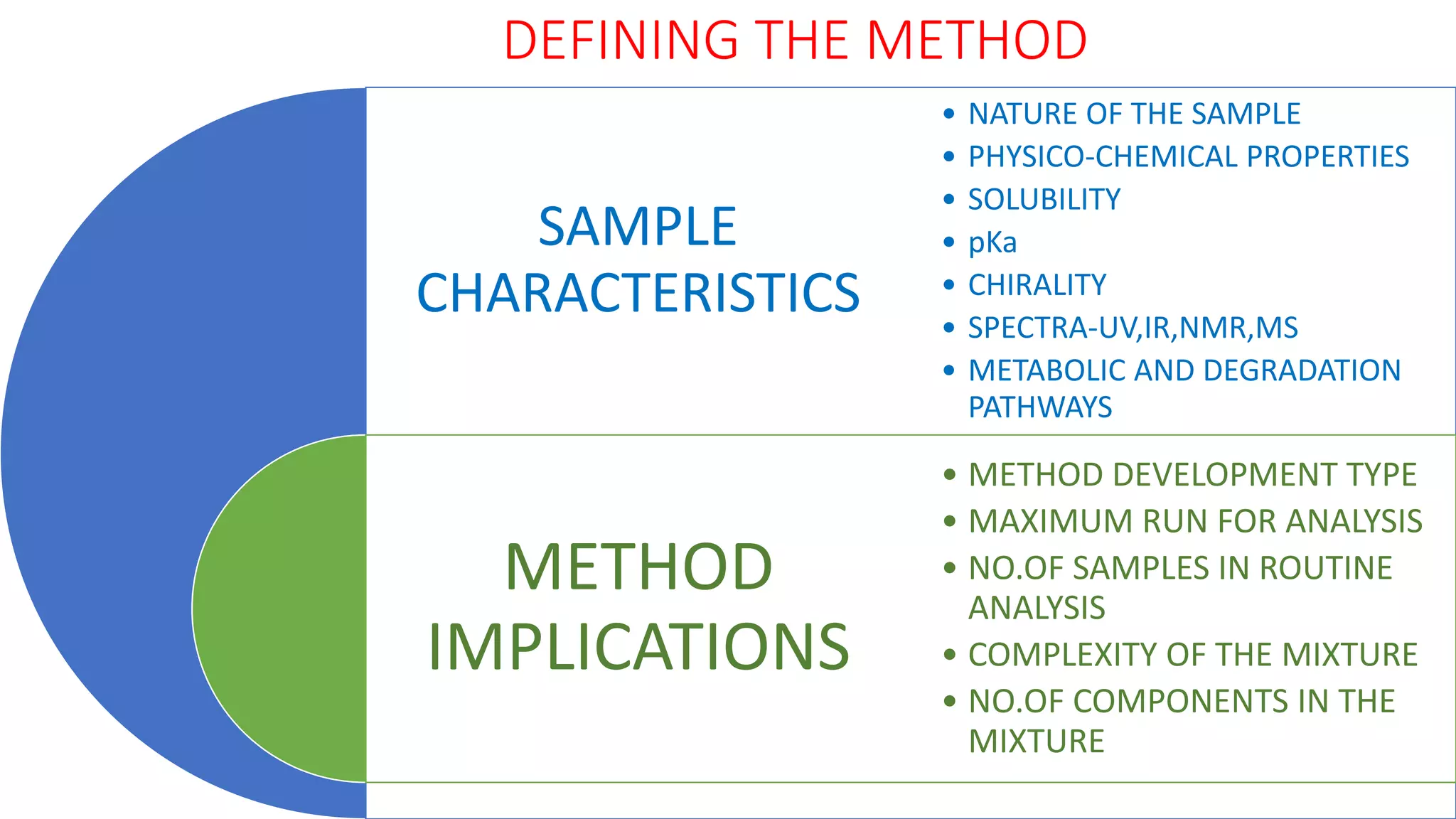
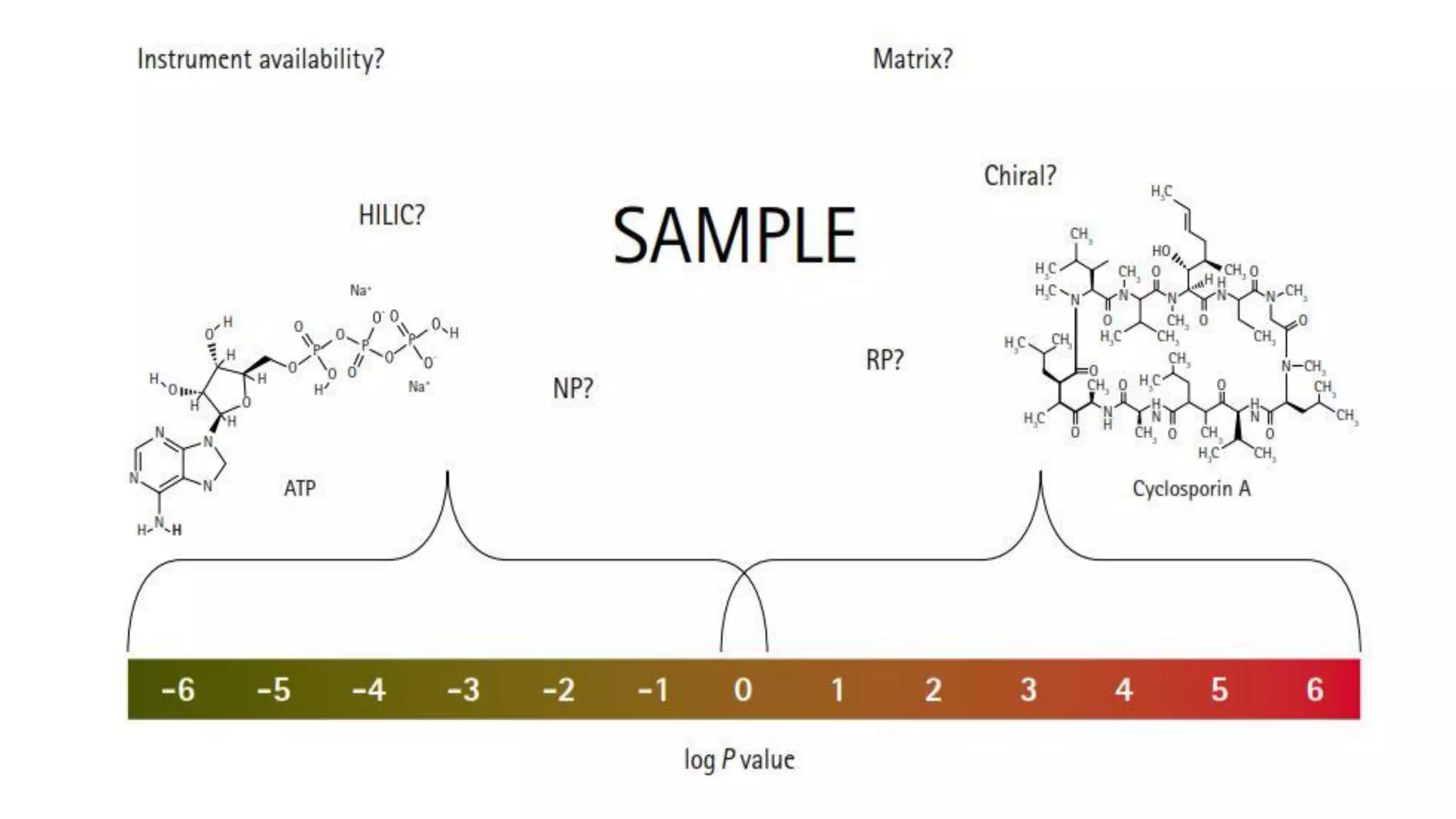







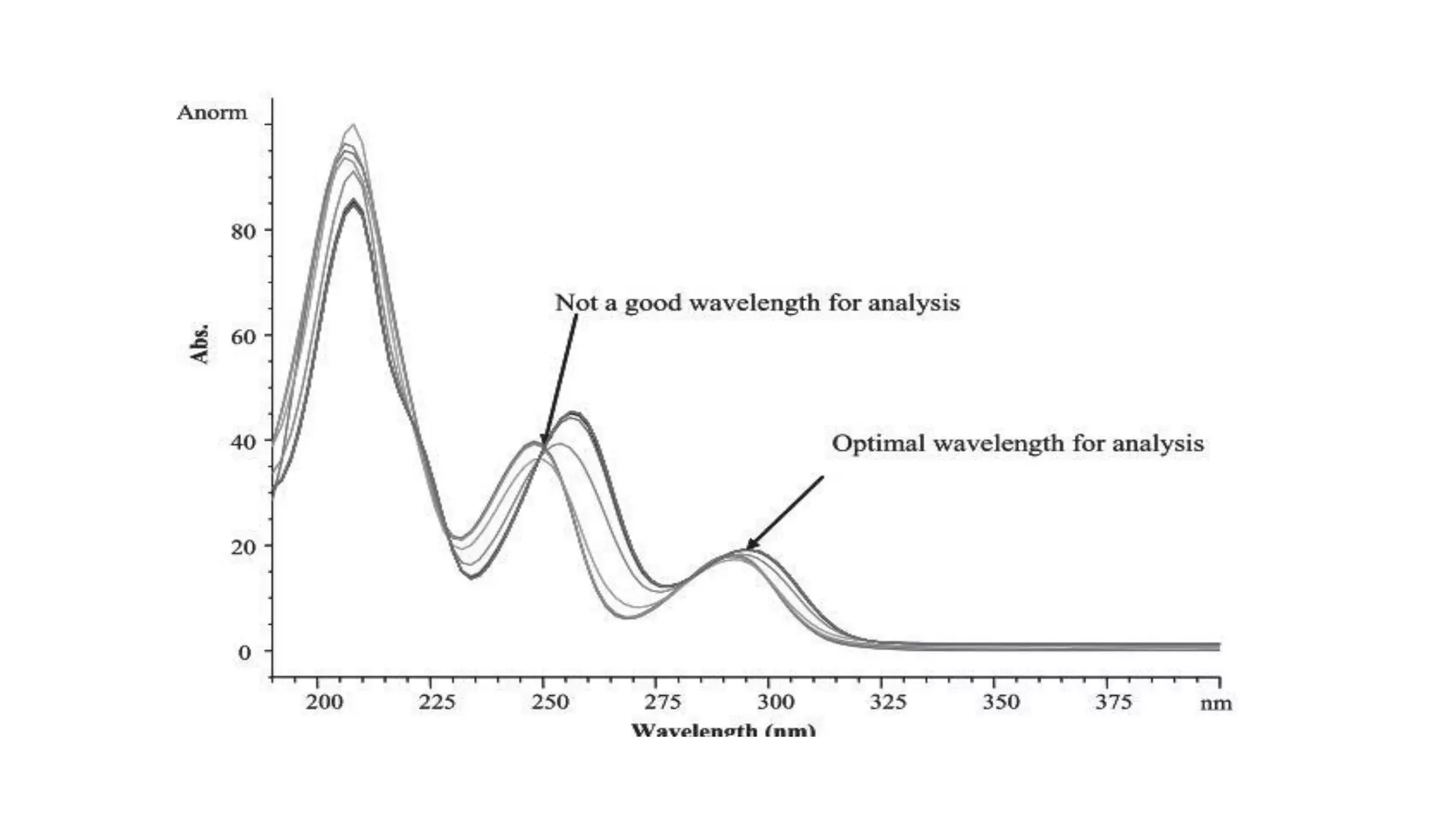













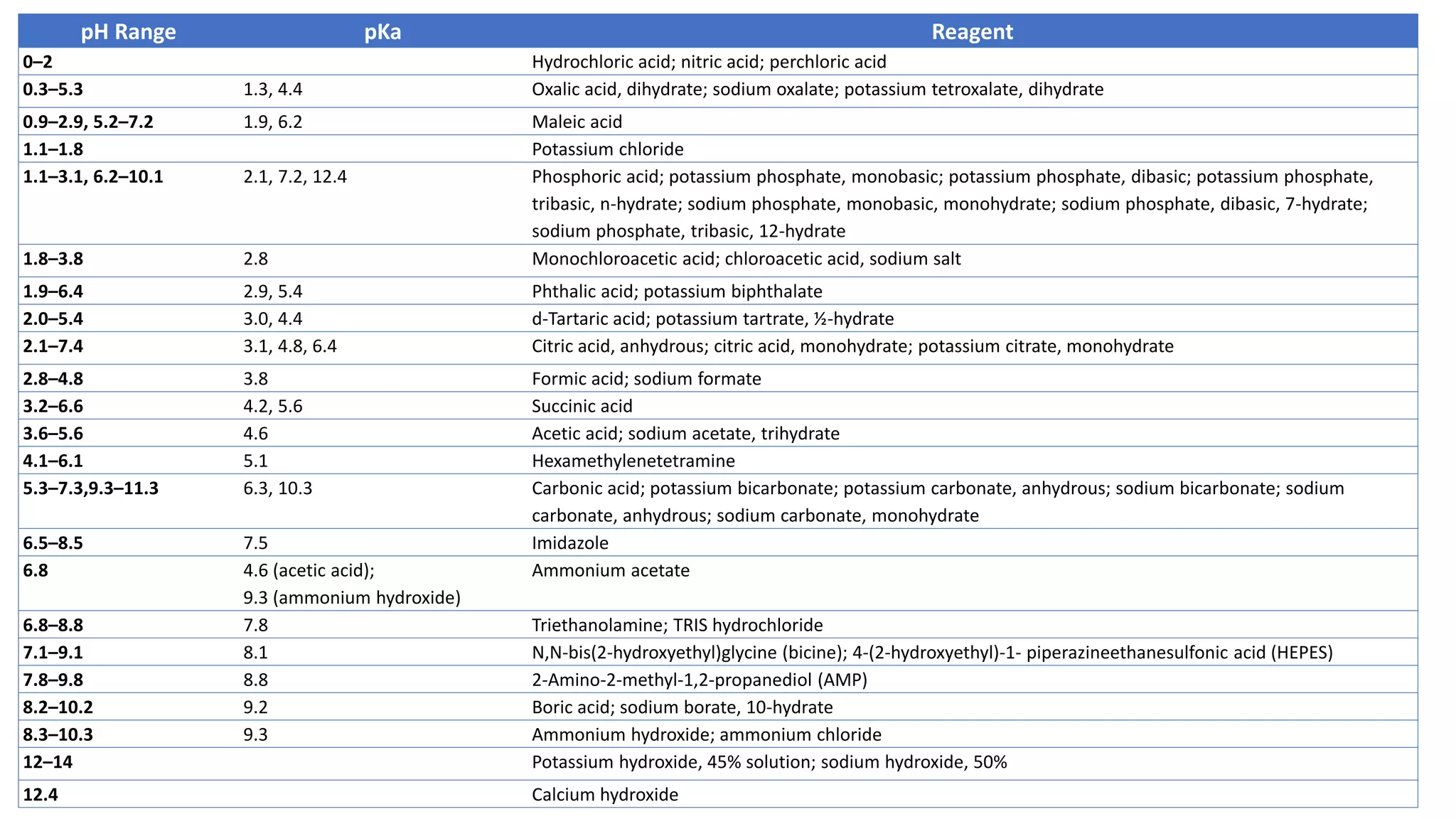







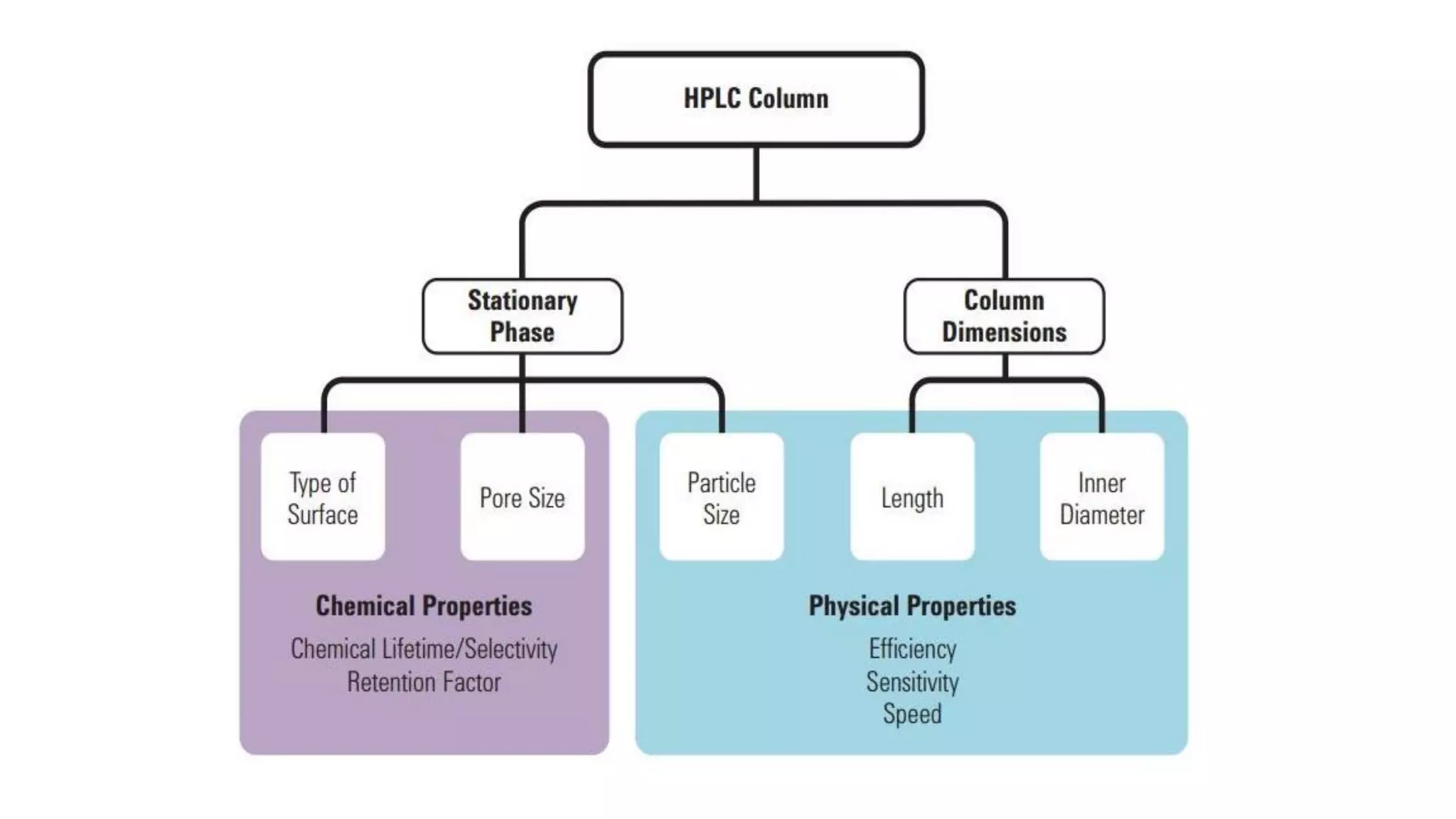

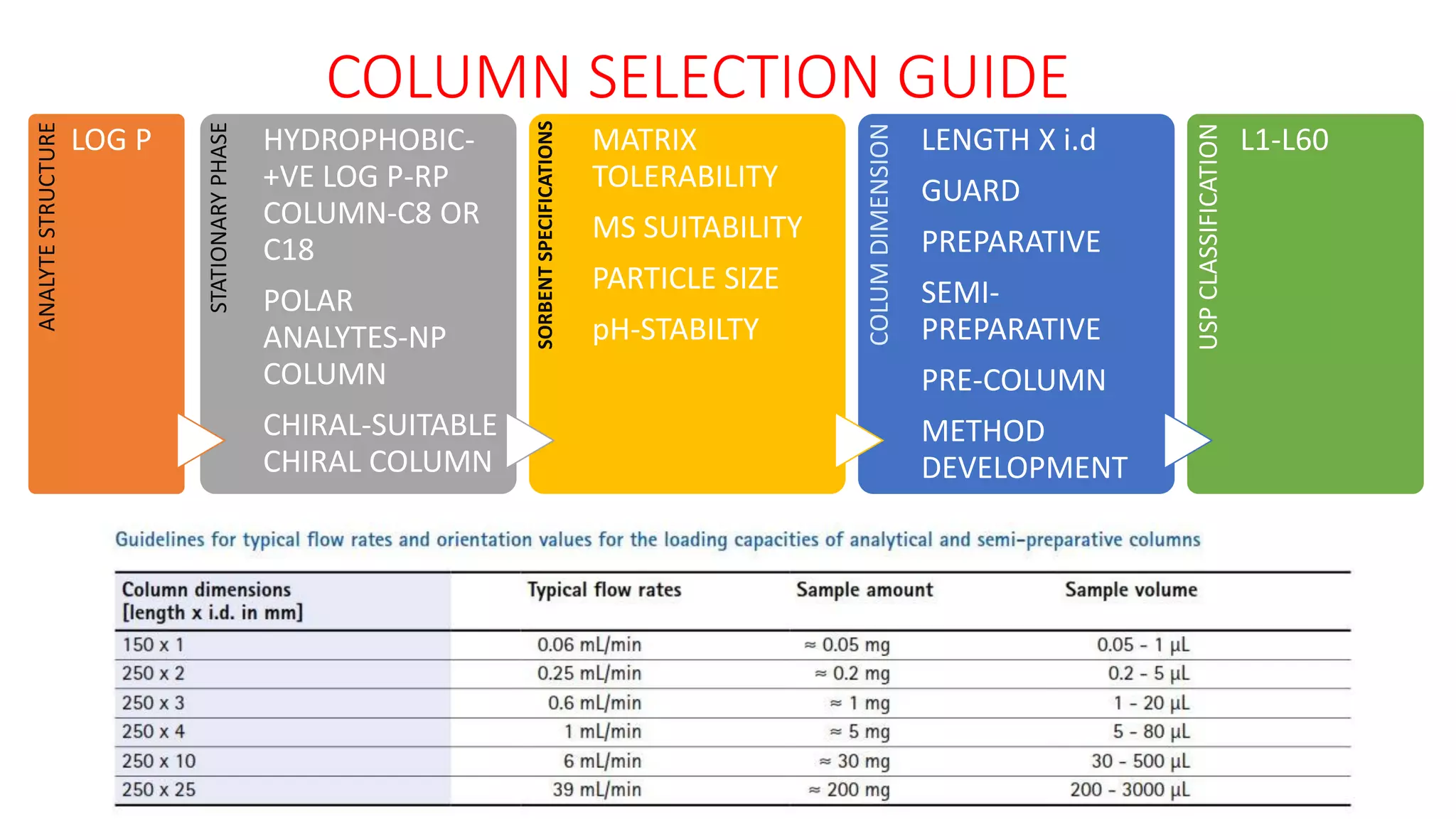






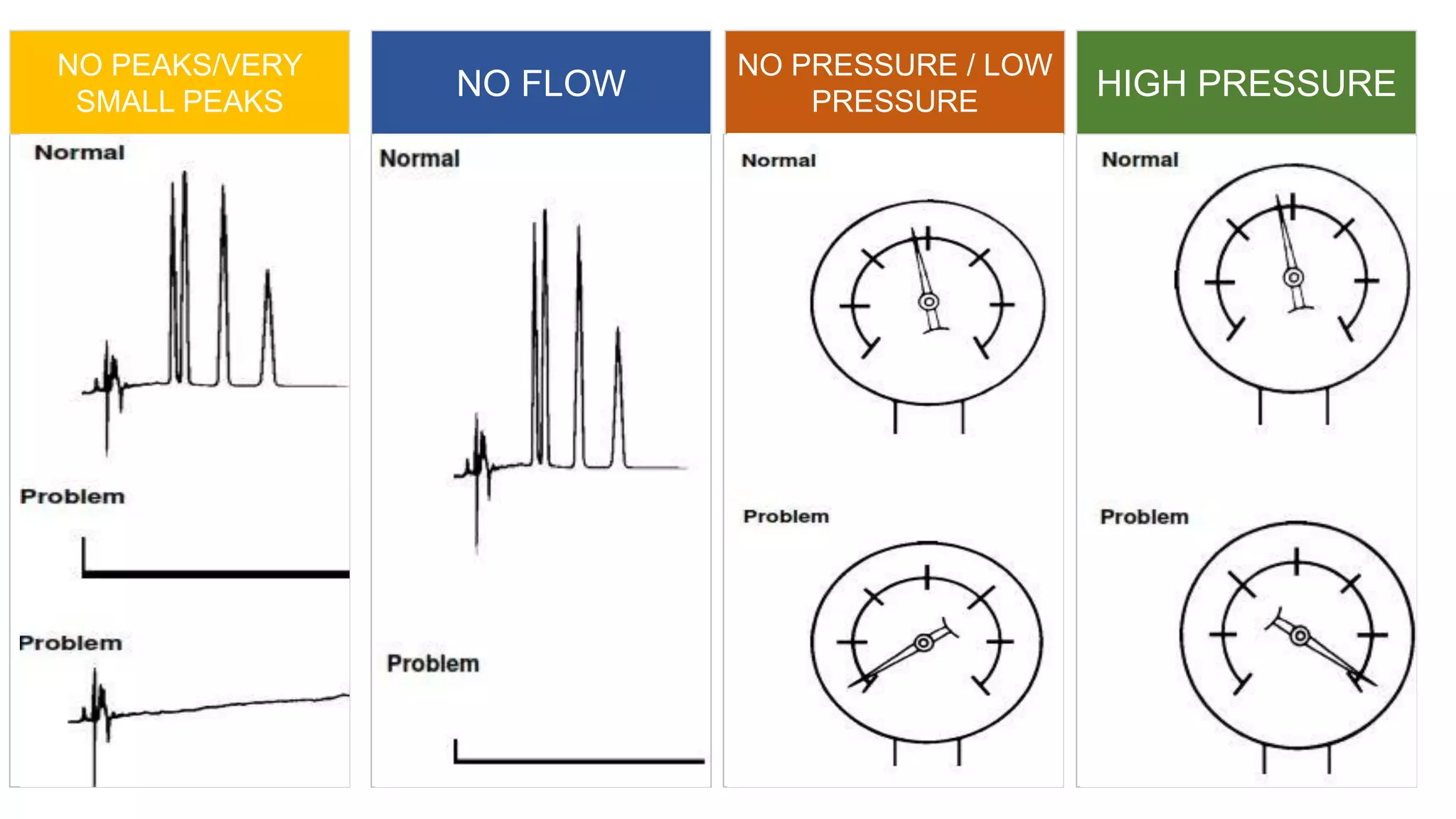


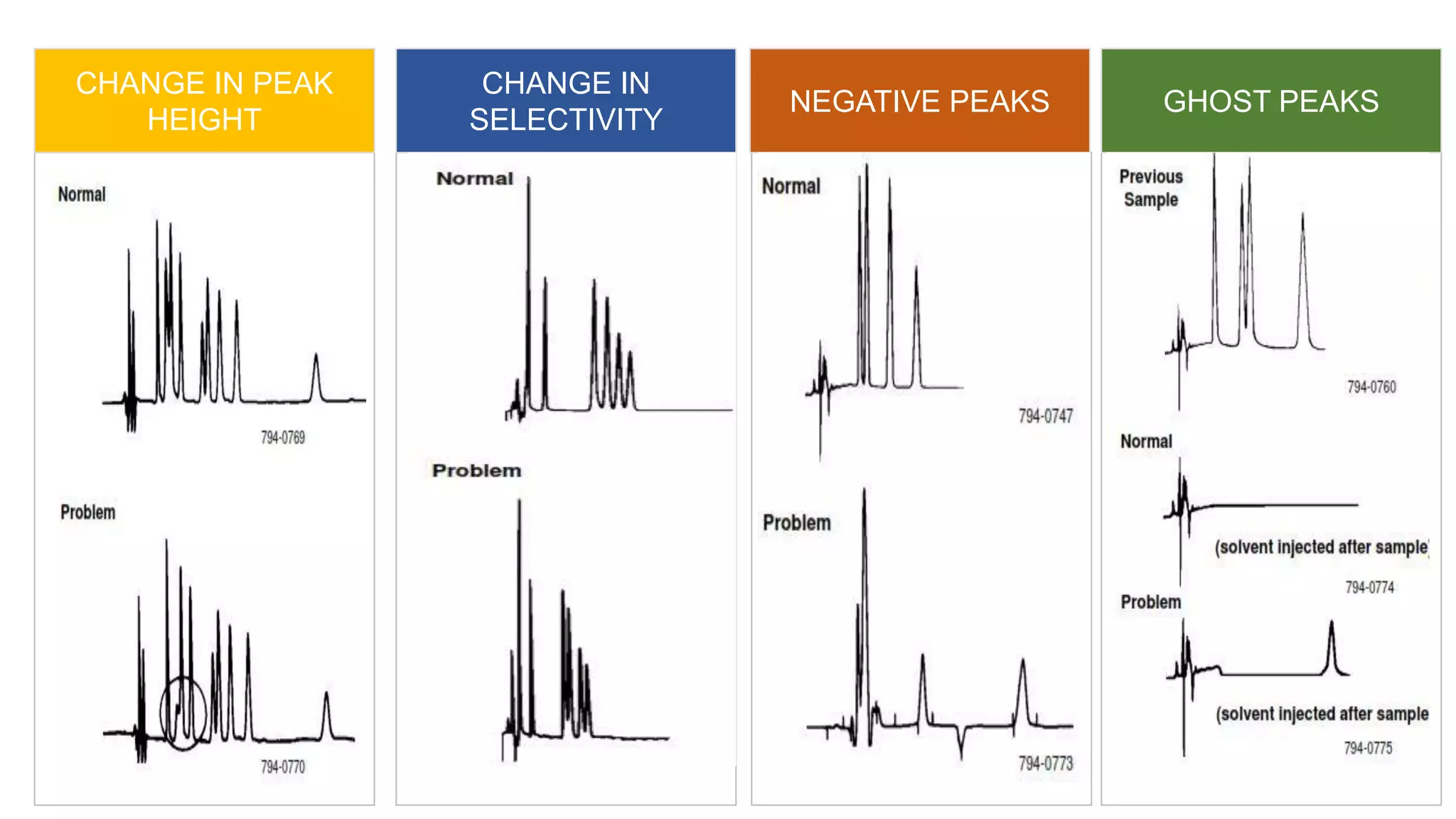







The document outlines the development of High-Performance Liquid Chromatography (HPLC) methods, covering various types of HPLC, instrumentation, and important parameters like retention time and separation factors. It emphasizes method development as both an art and a science, noting common mistakes and essential protocol steps, including sample preparation and mobile phase selection. Key considerations for optimally choosing buffers, detectors, and modifiers are discussed, alongside practical tips for ensuring effective column selection and troubleshooting.



































































