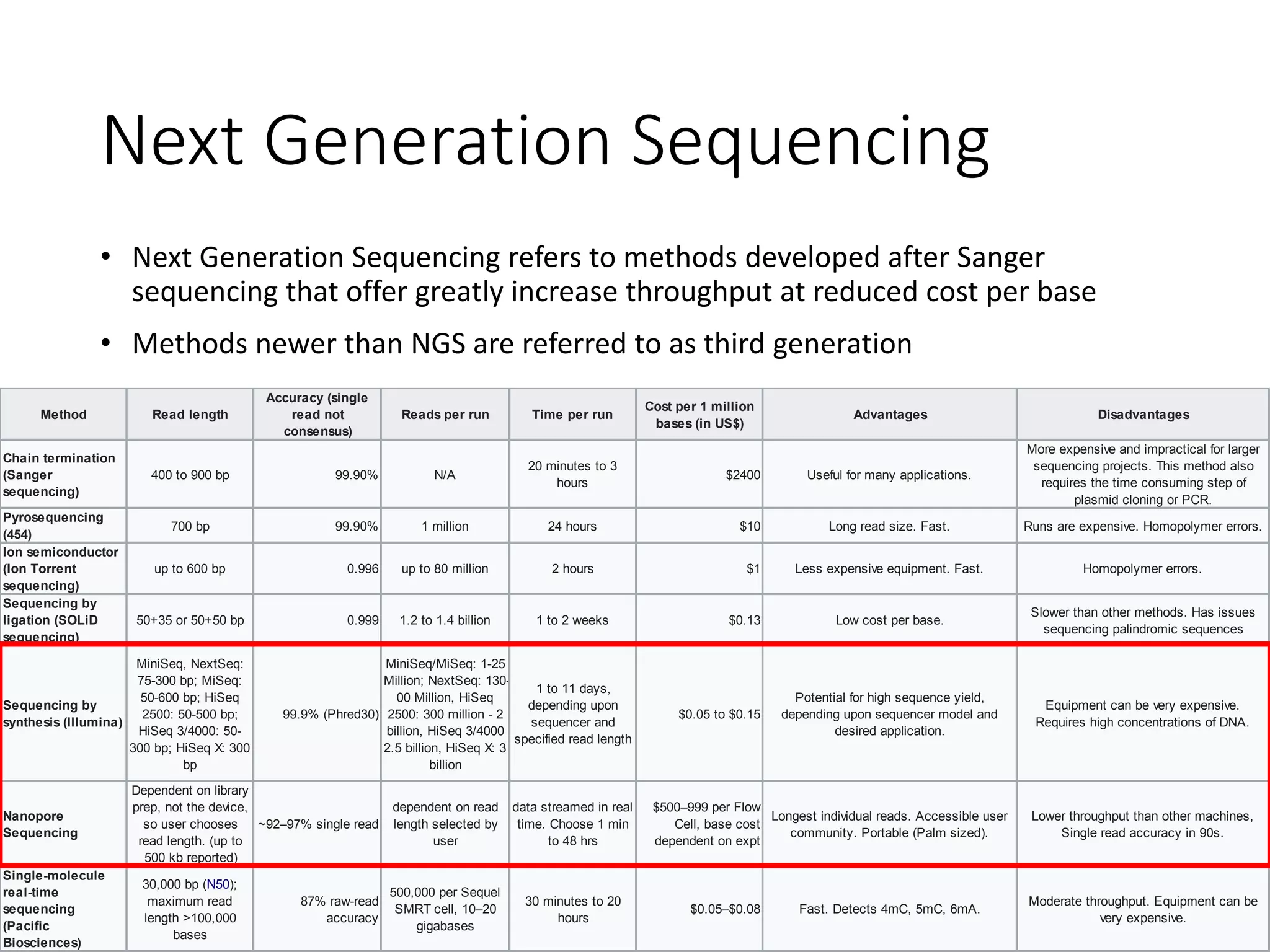
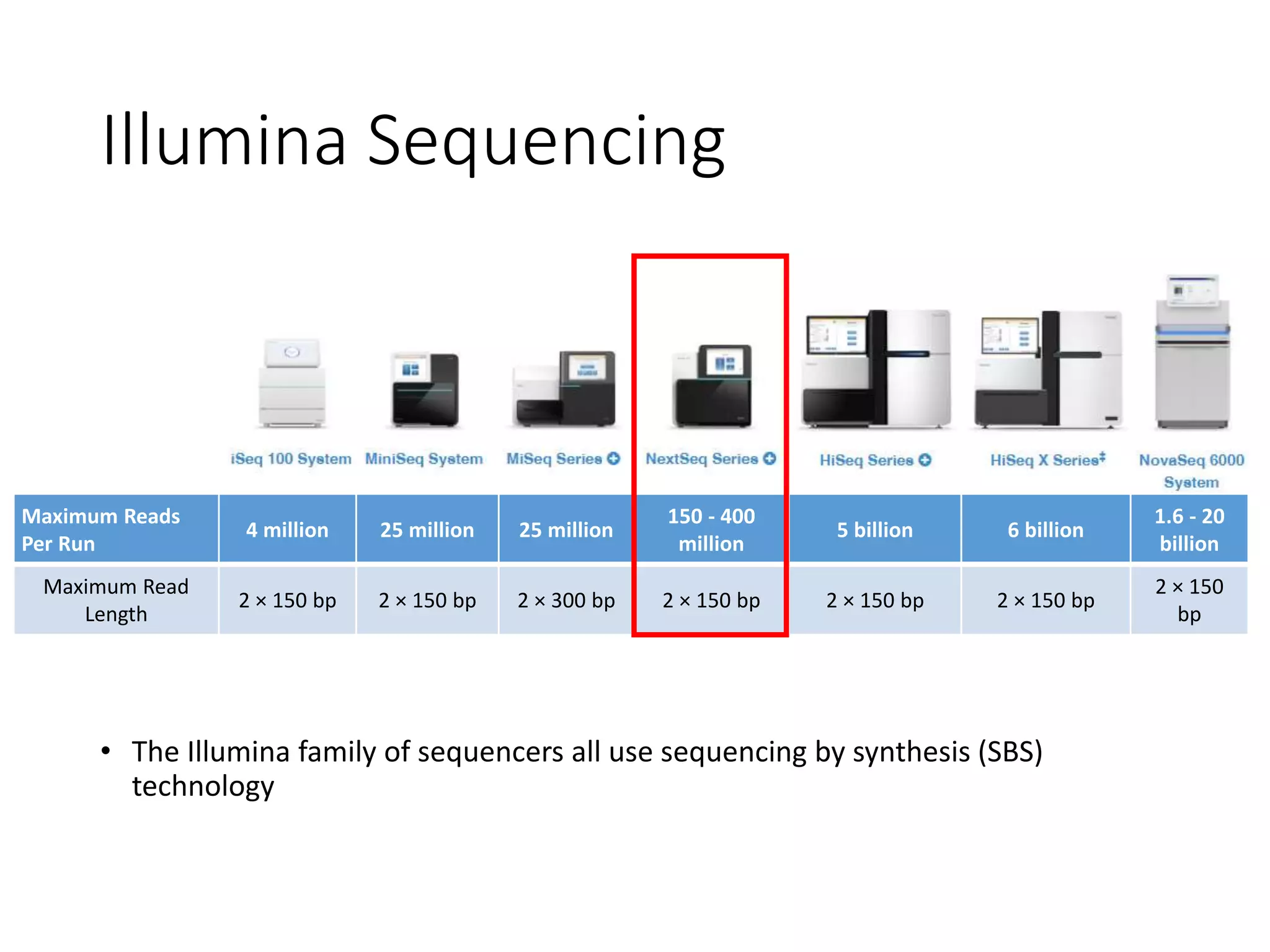
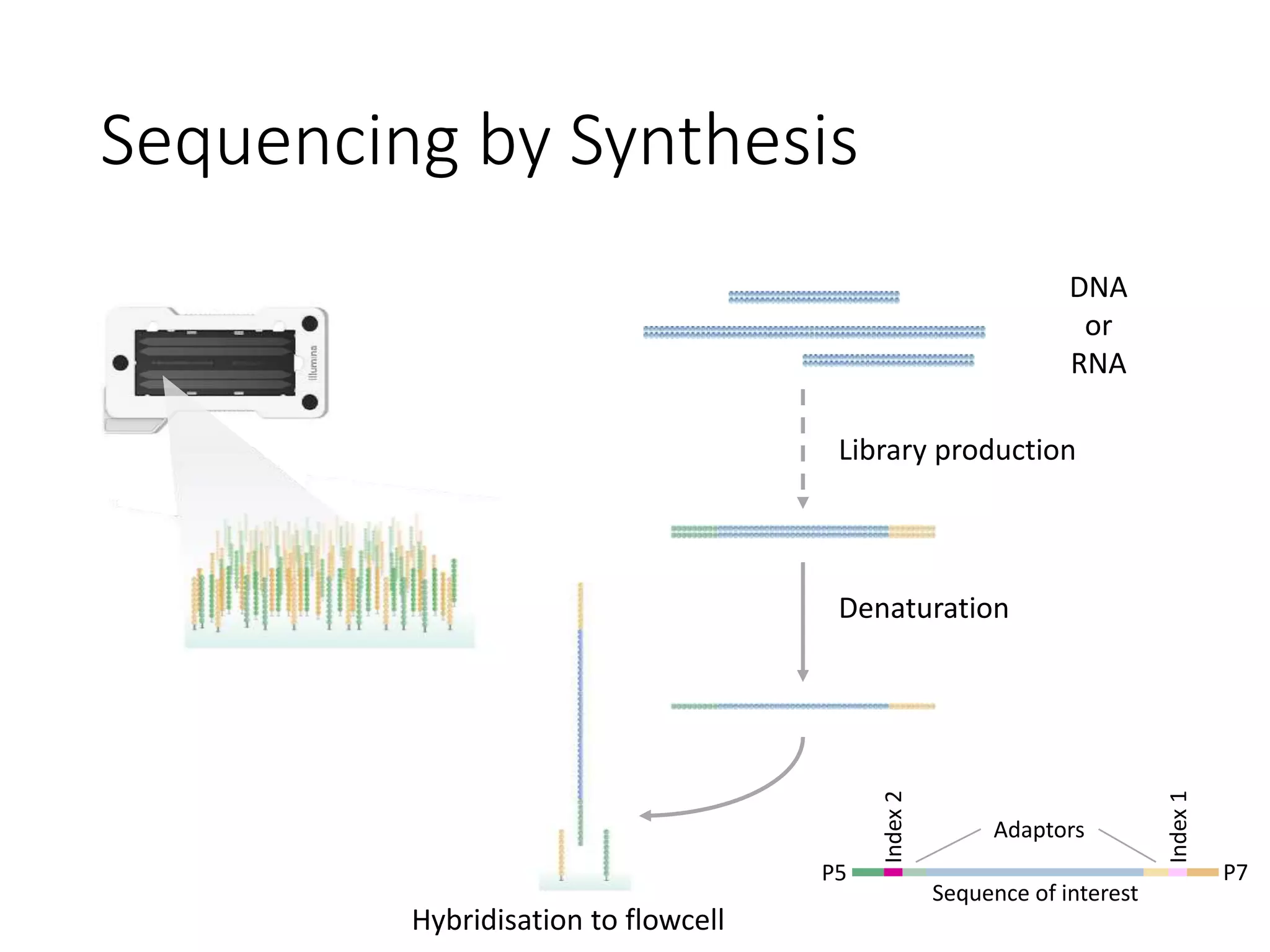
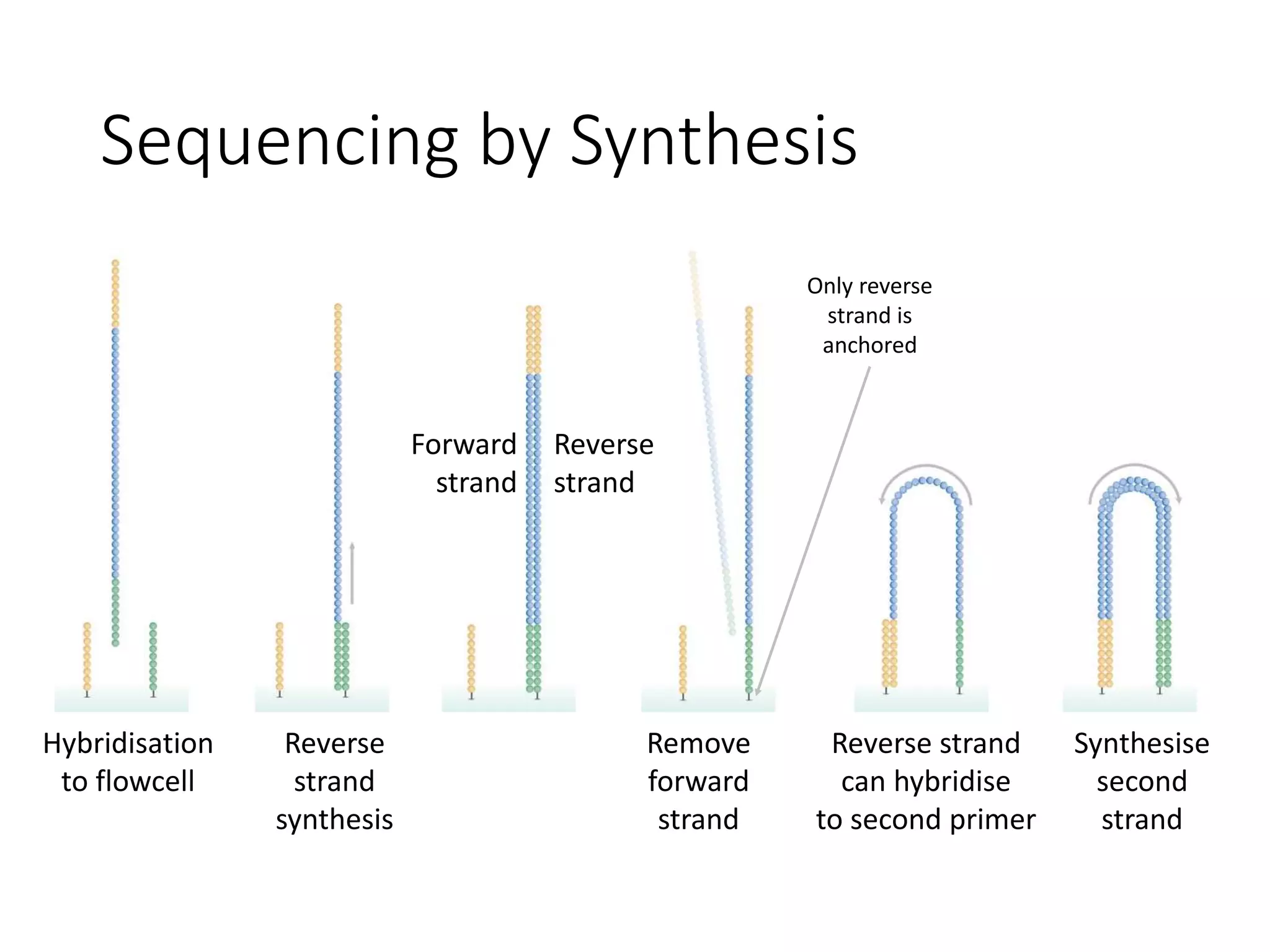
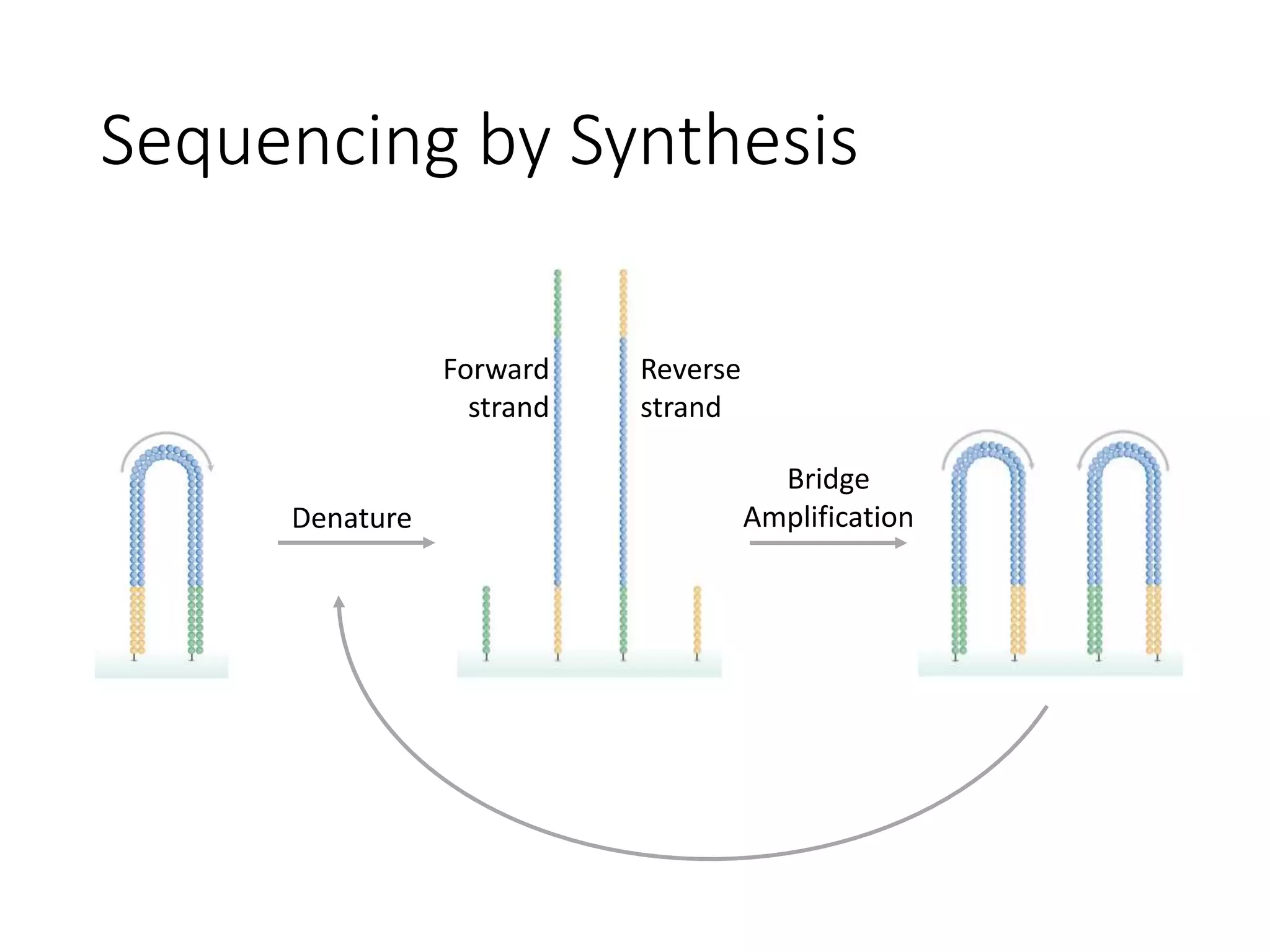
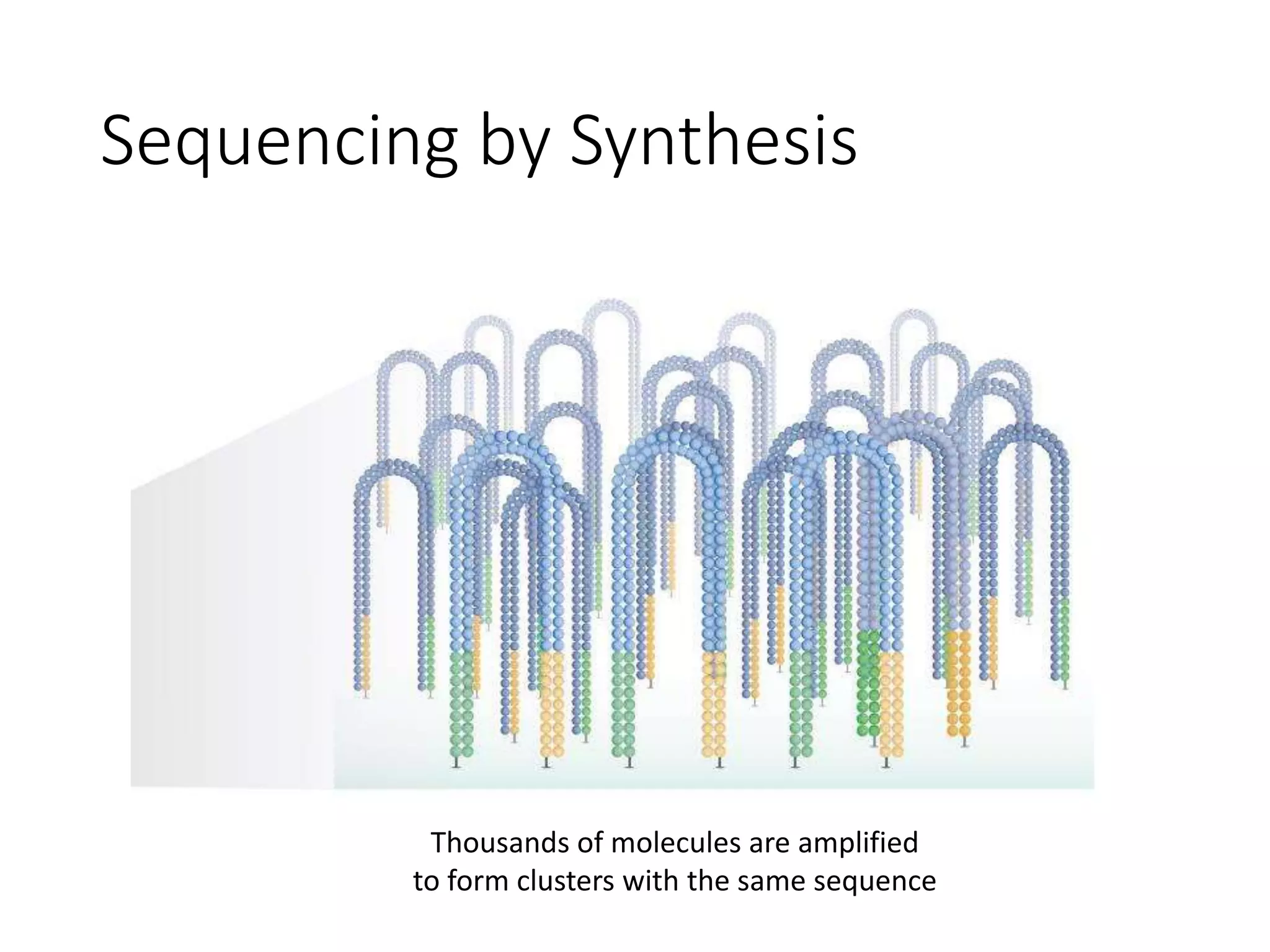
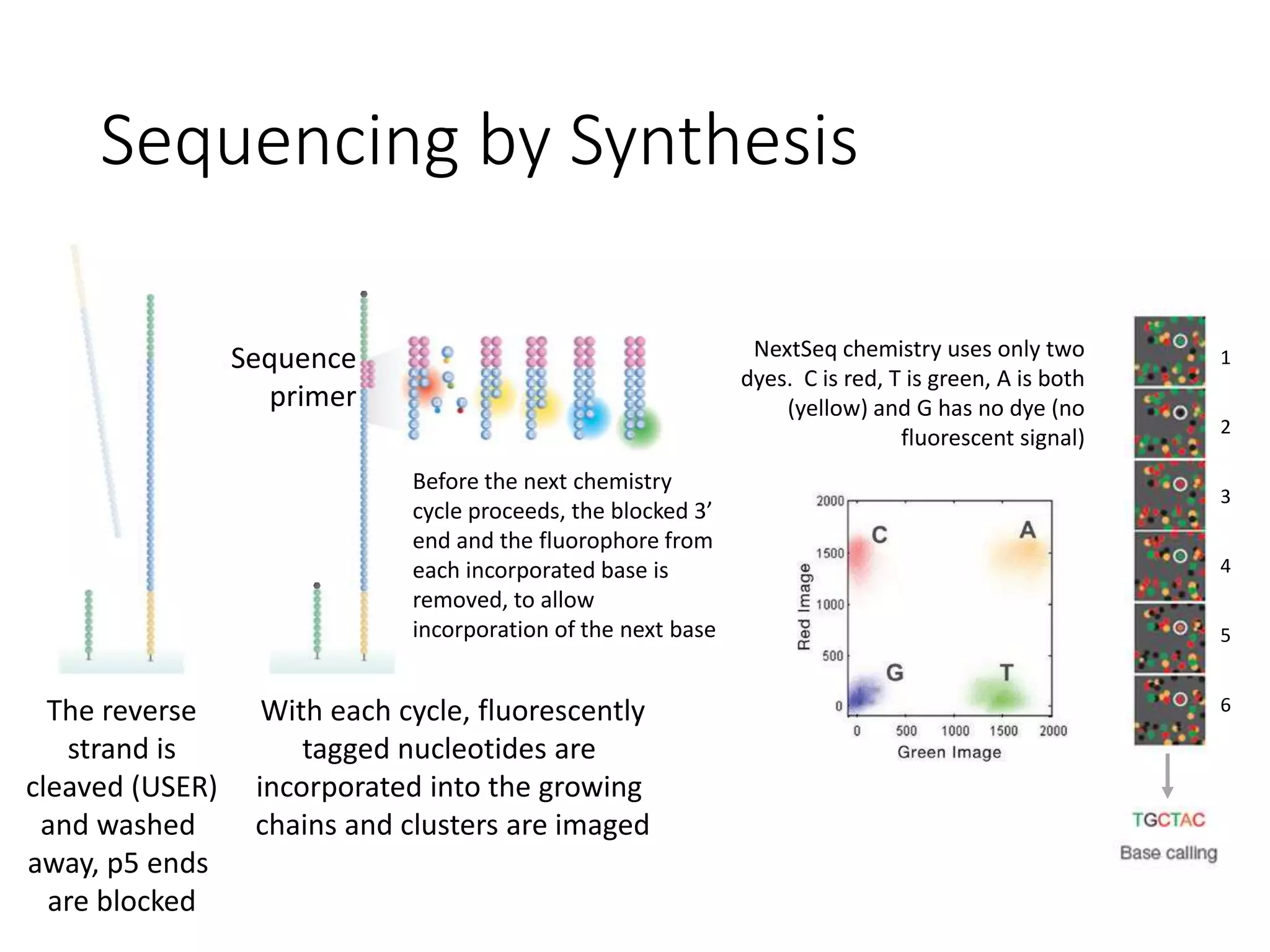
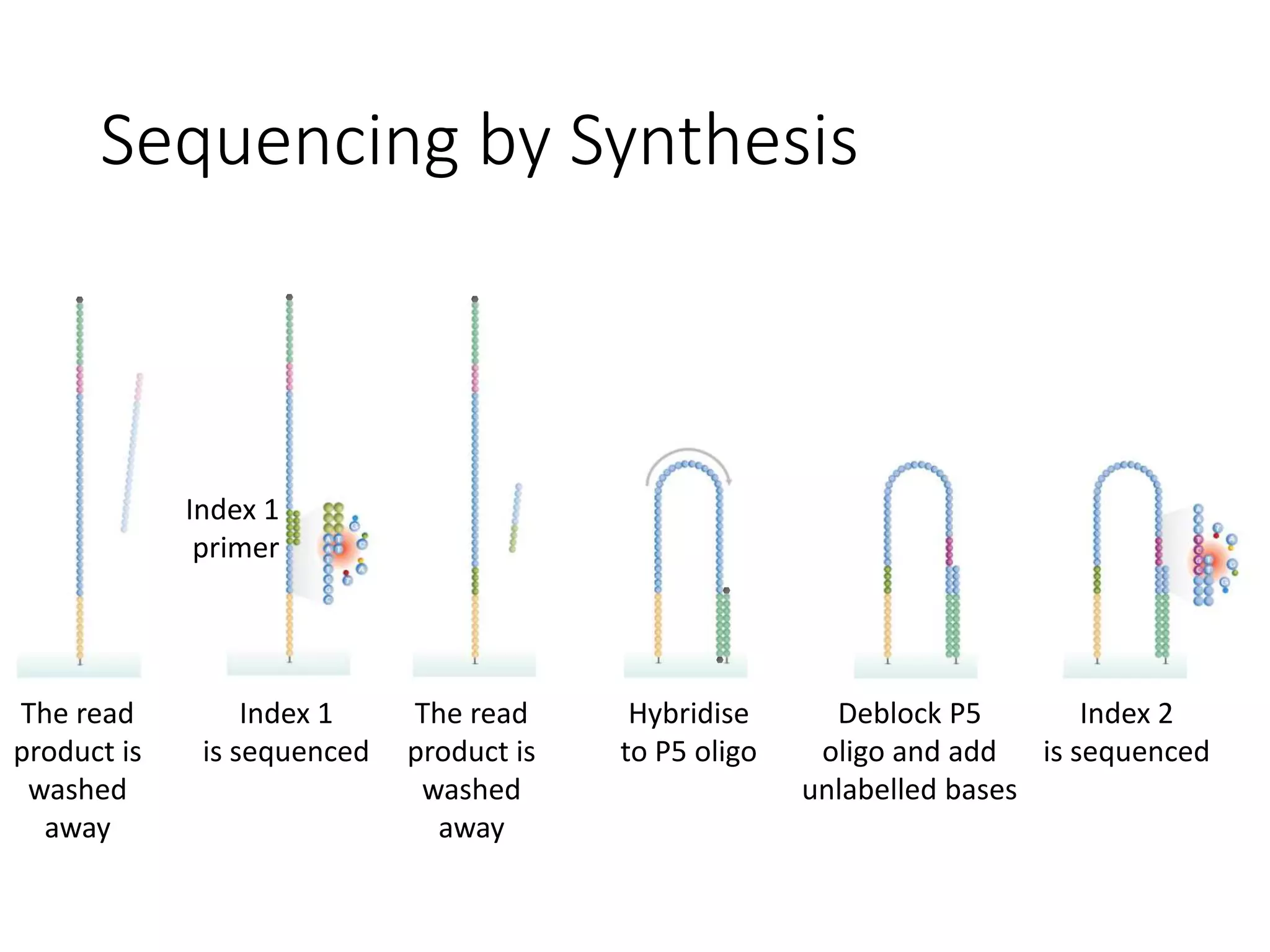
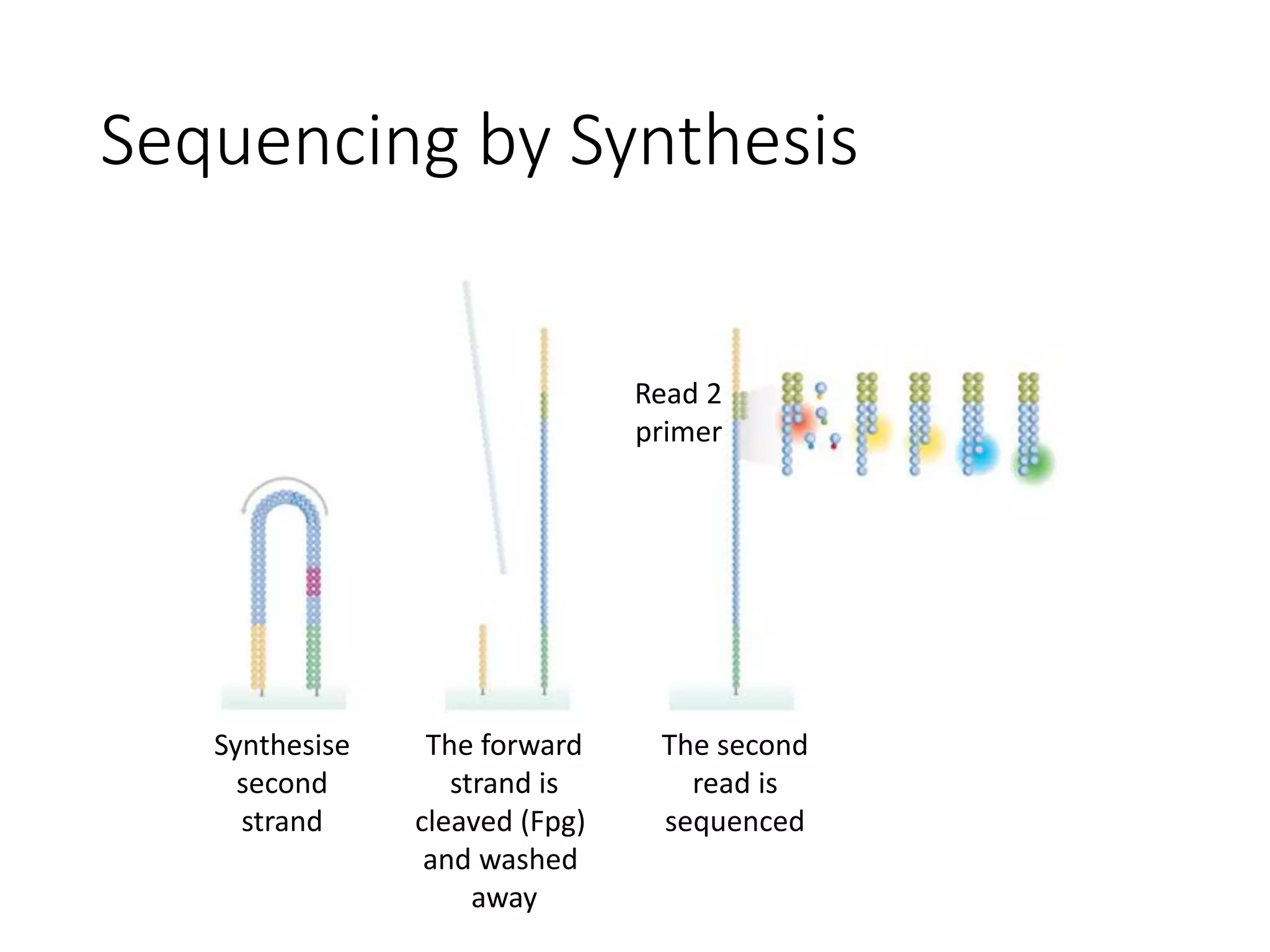
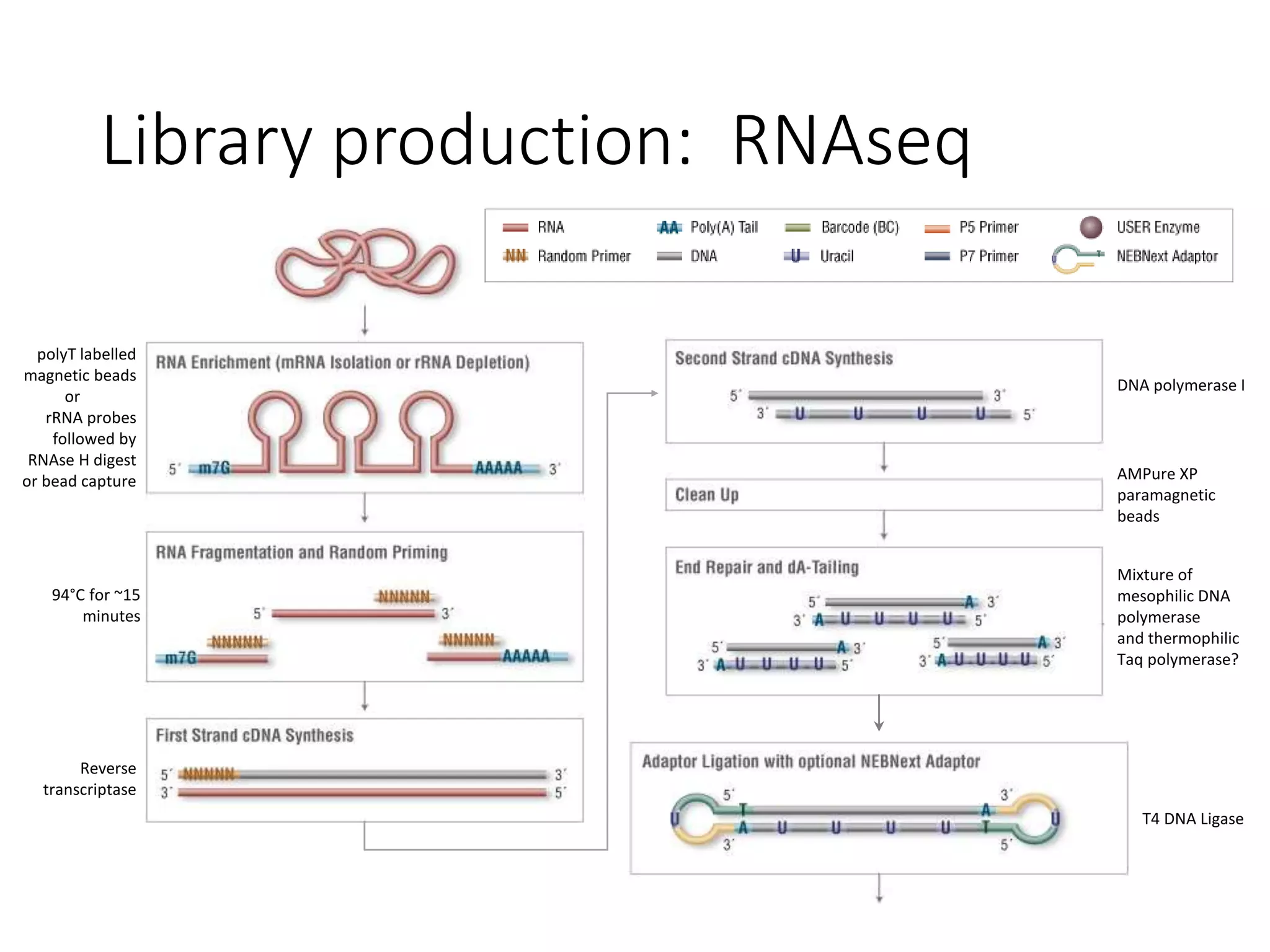
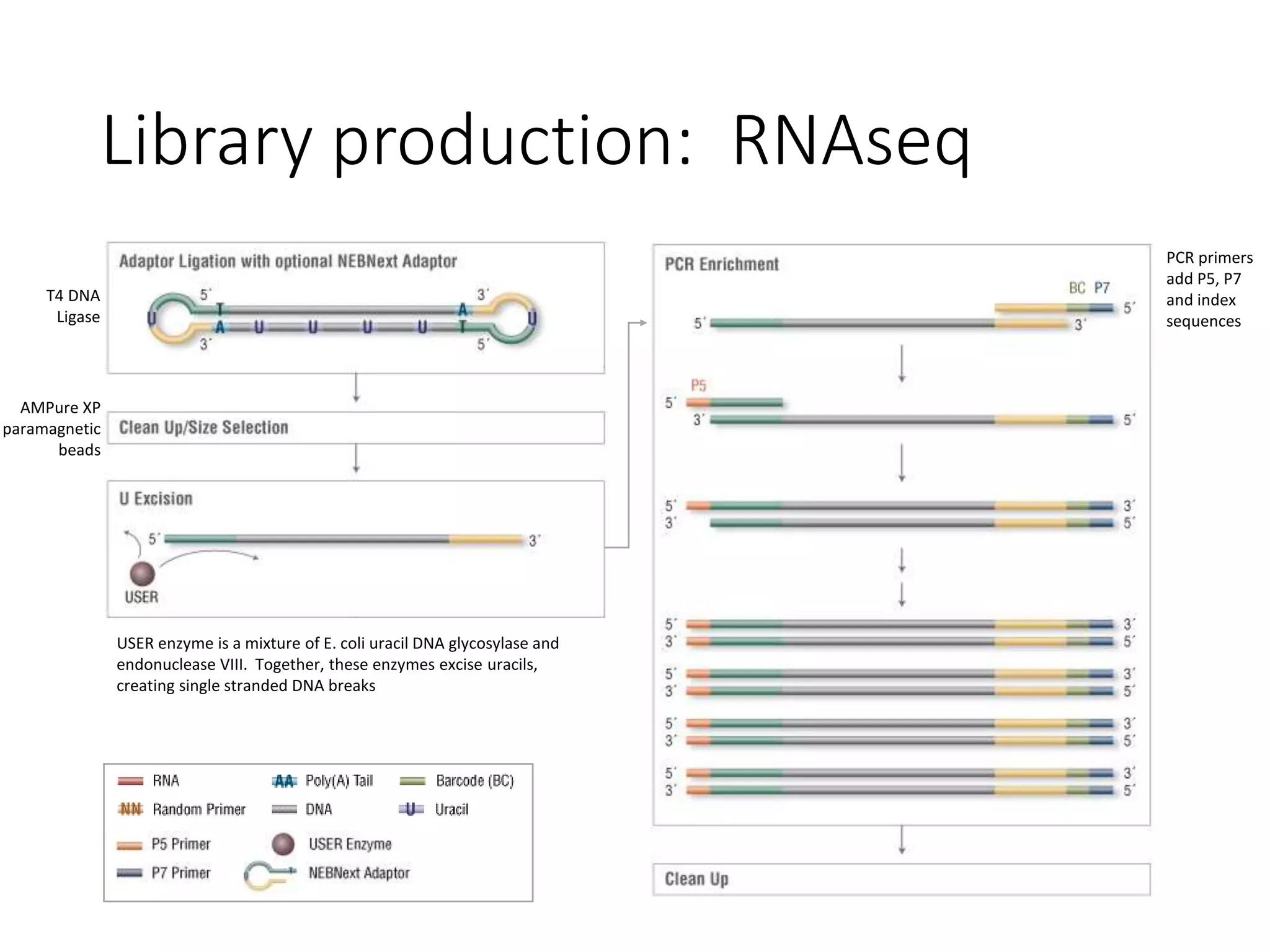
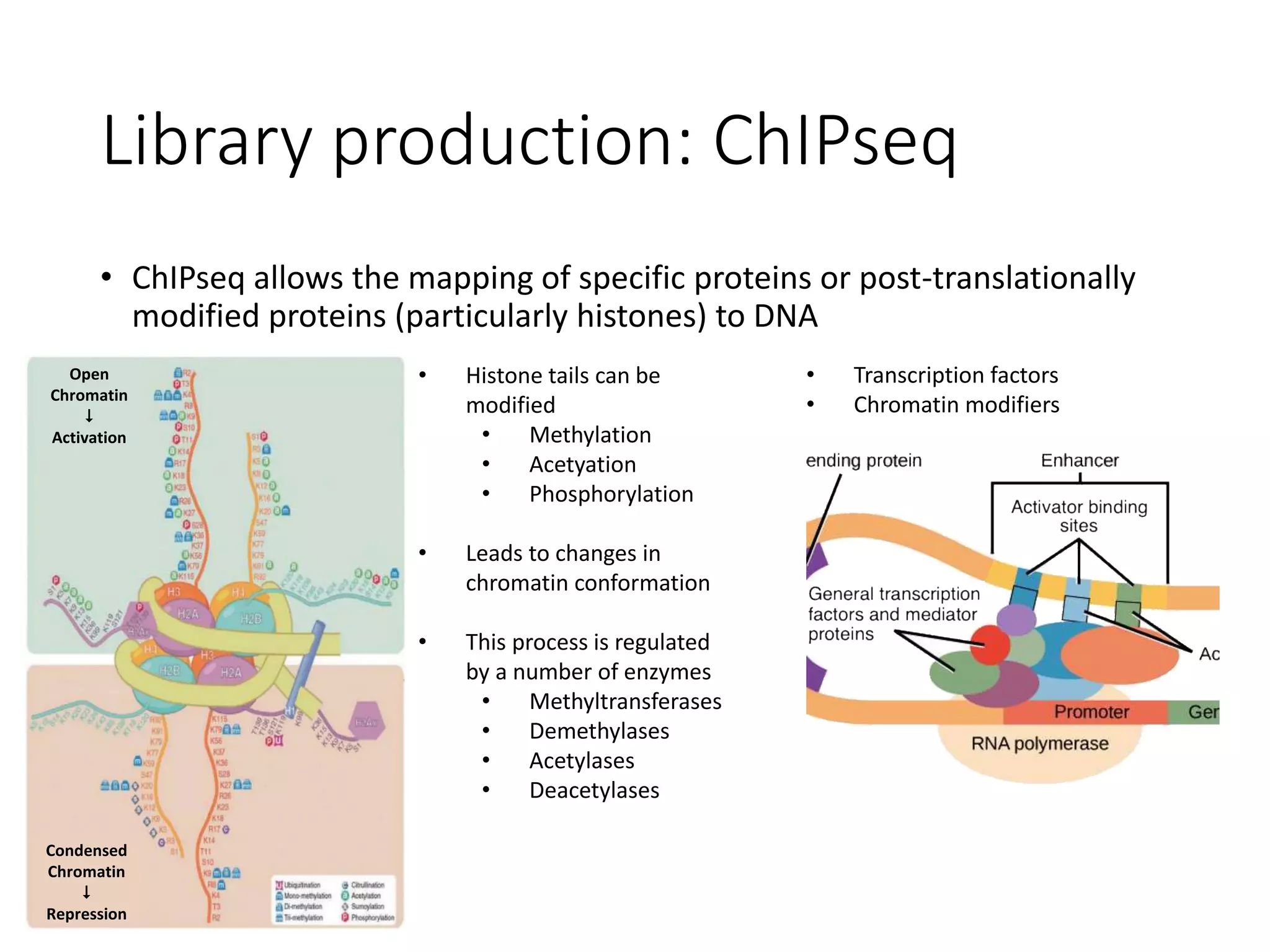
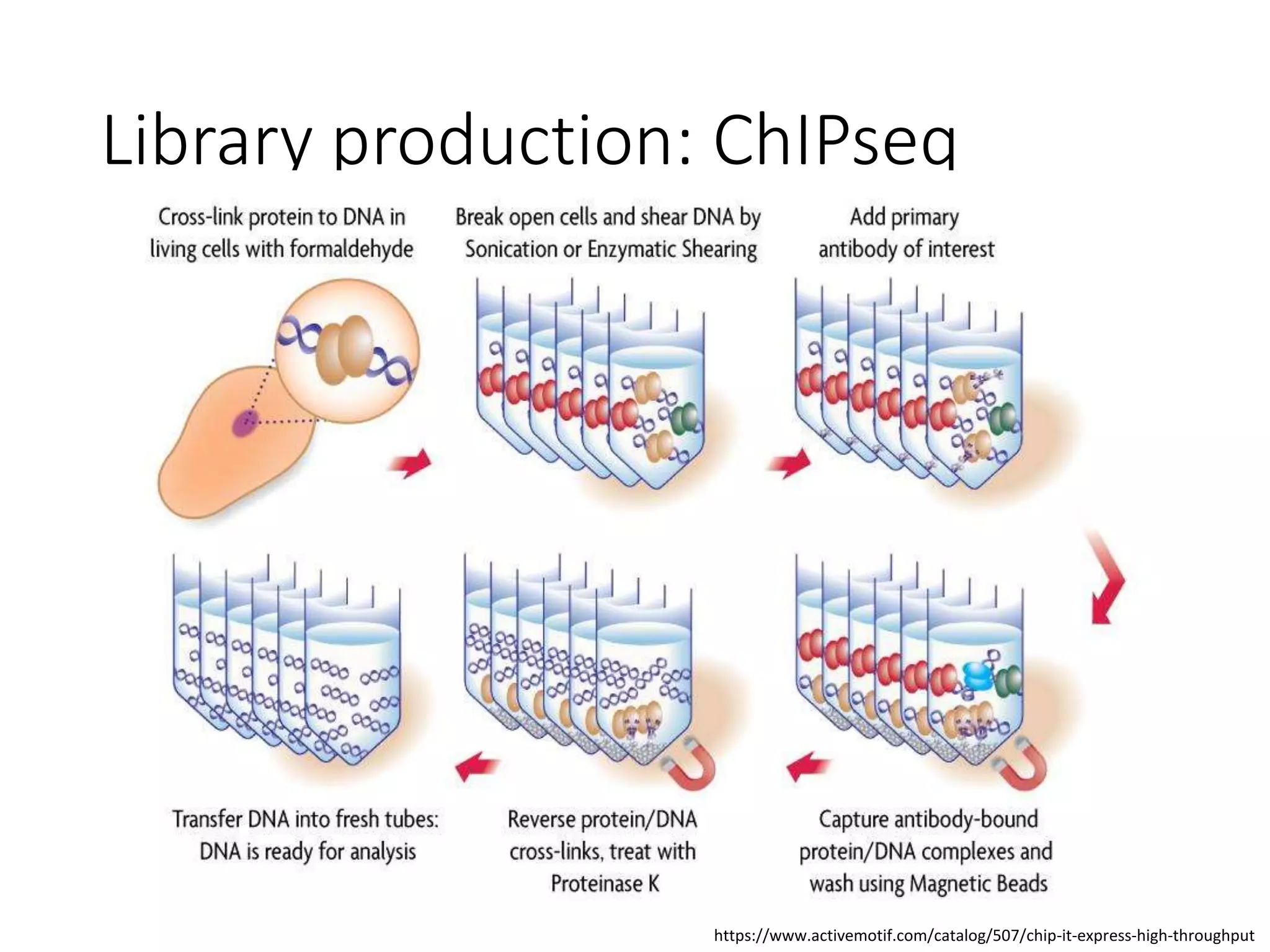
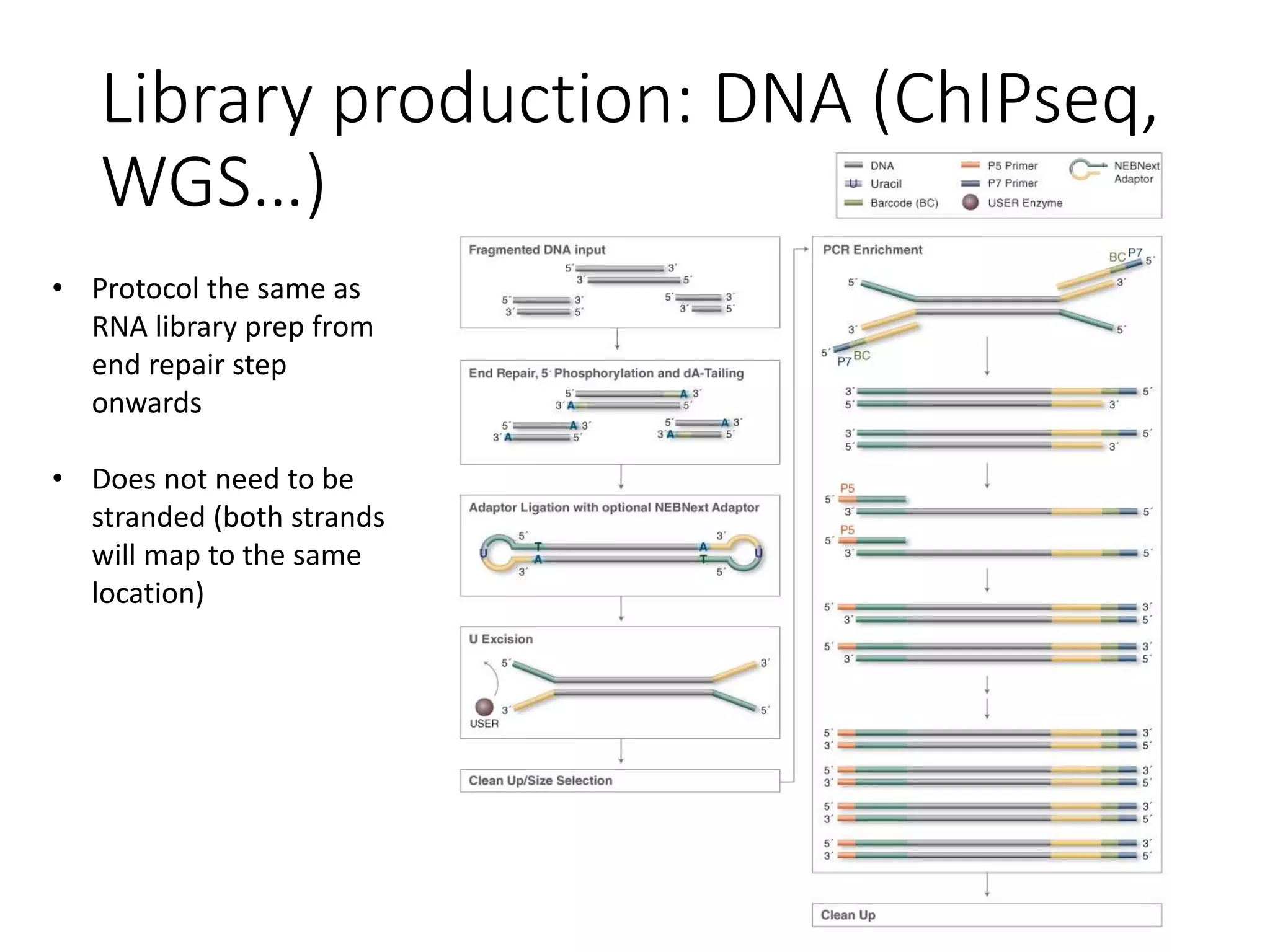
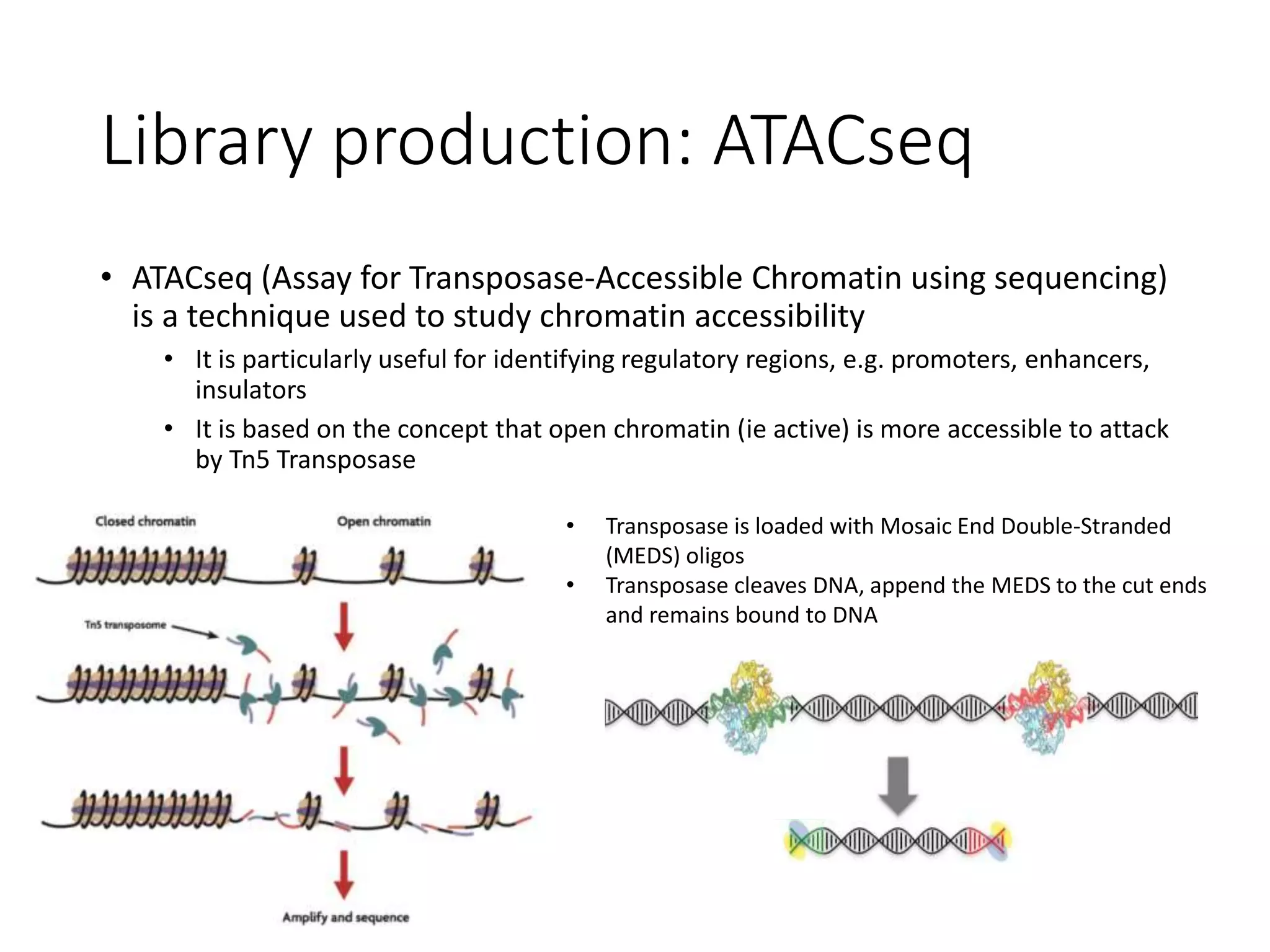
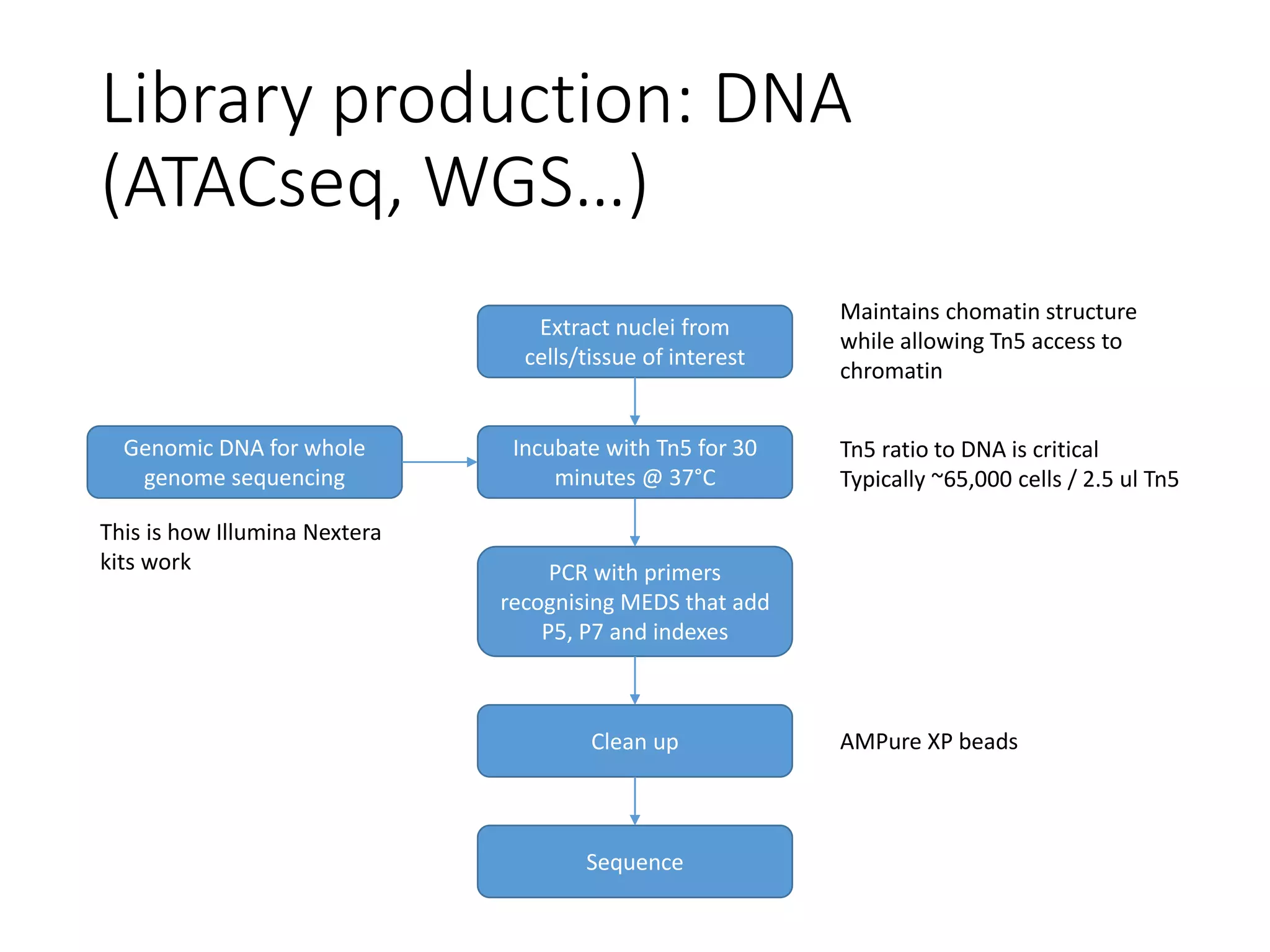
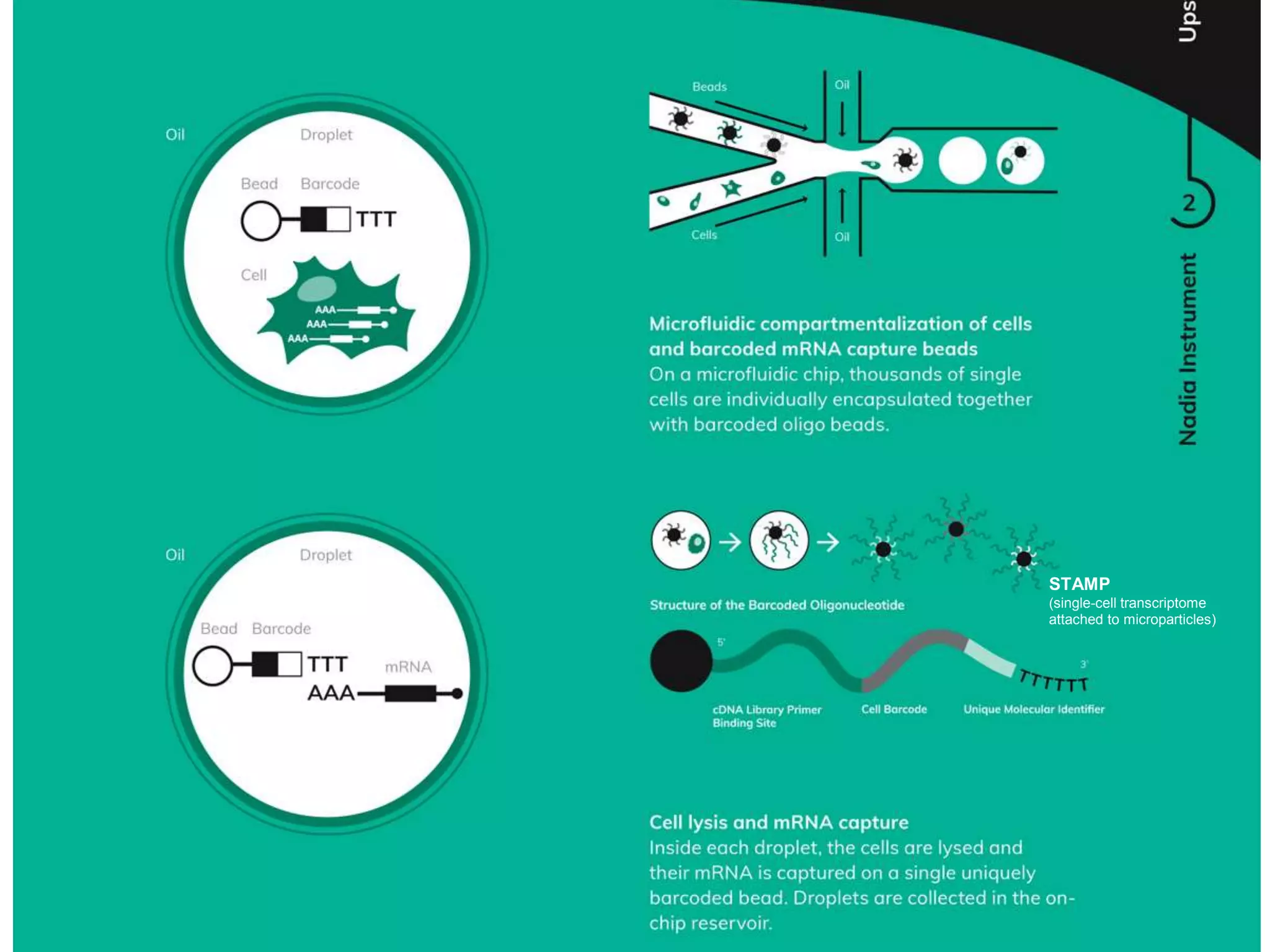
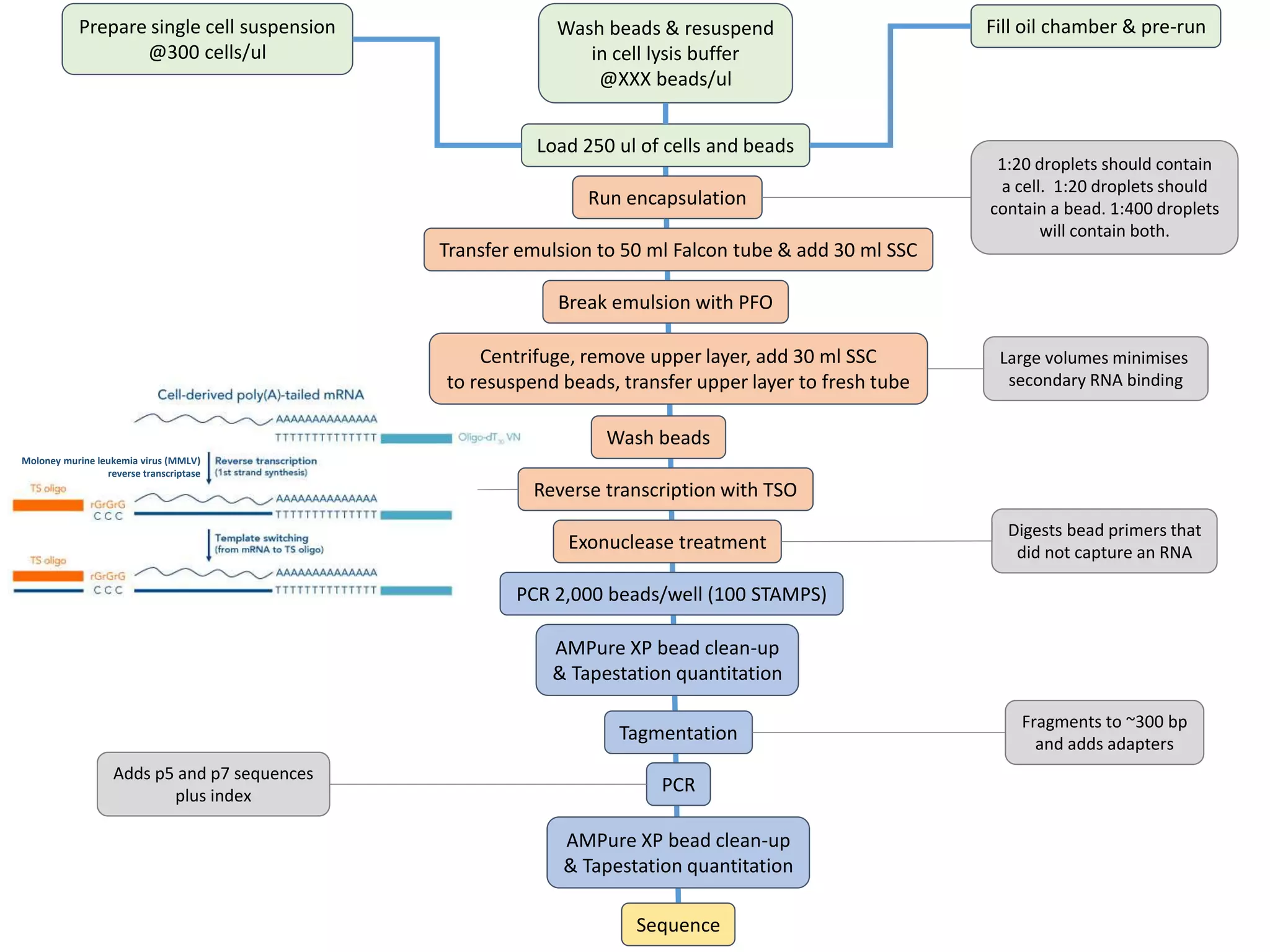
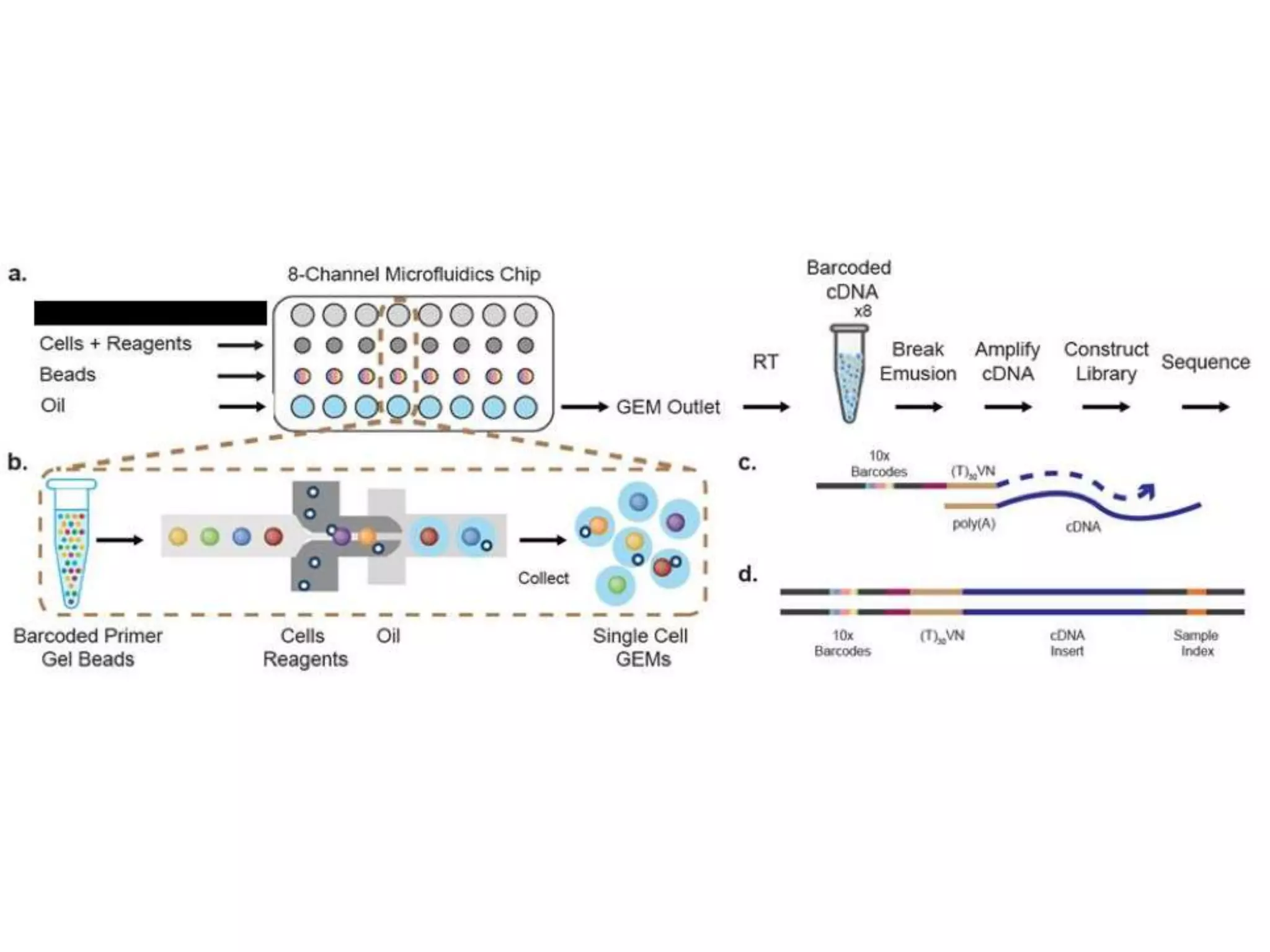
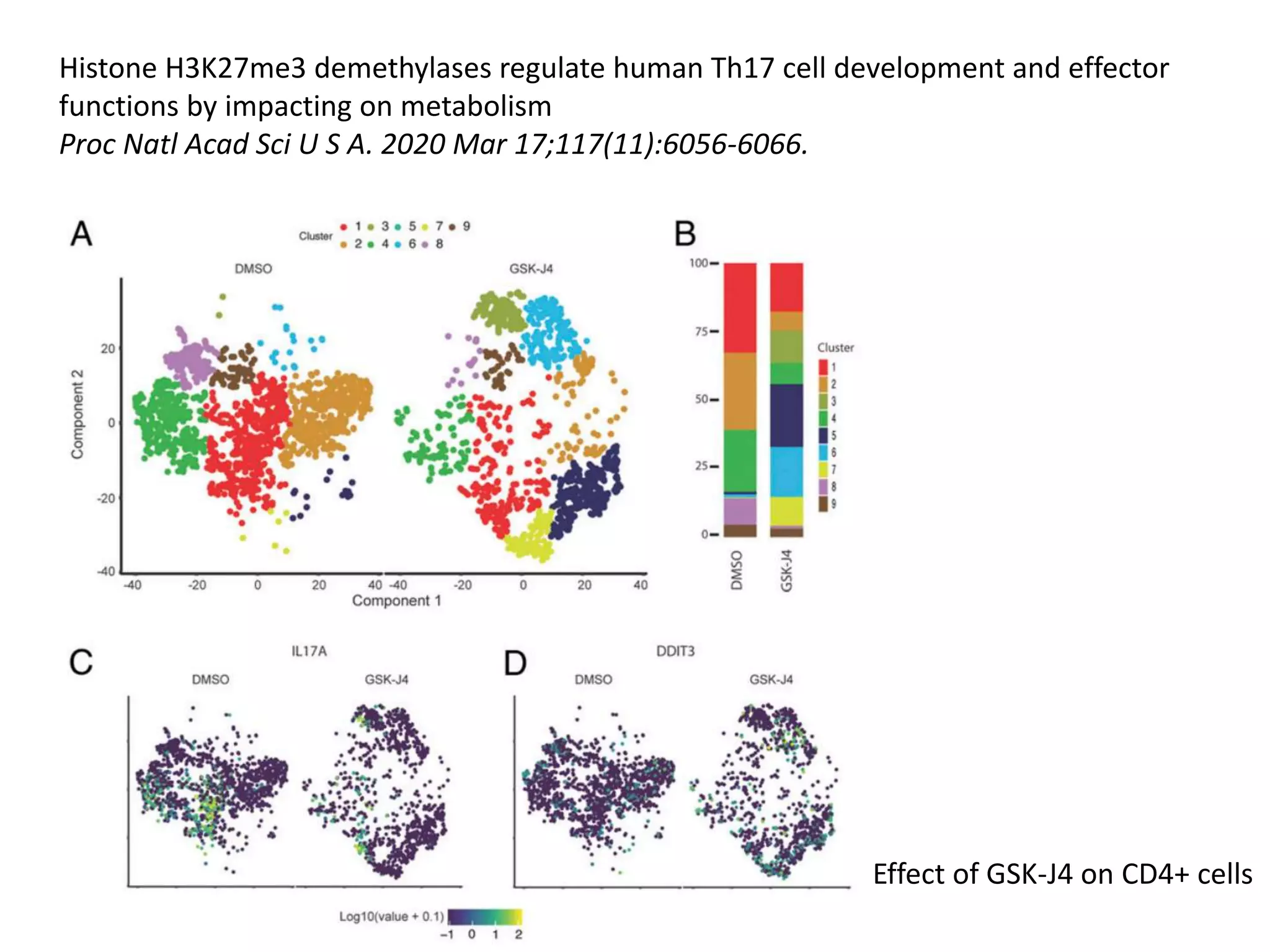
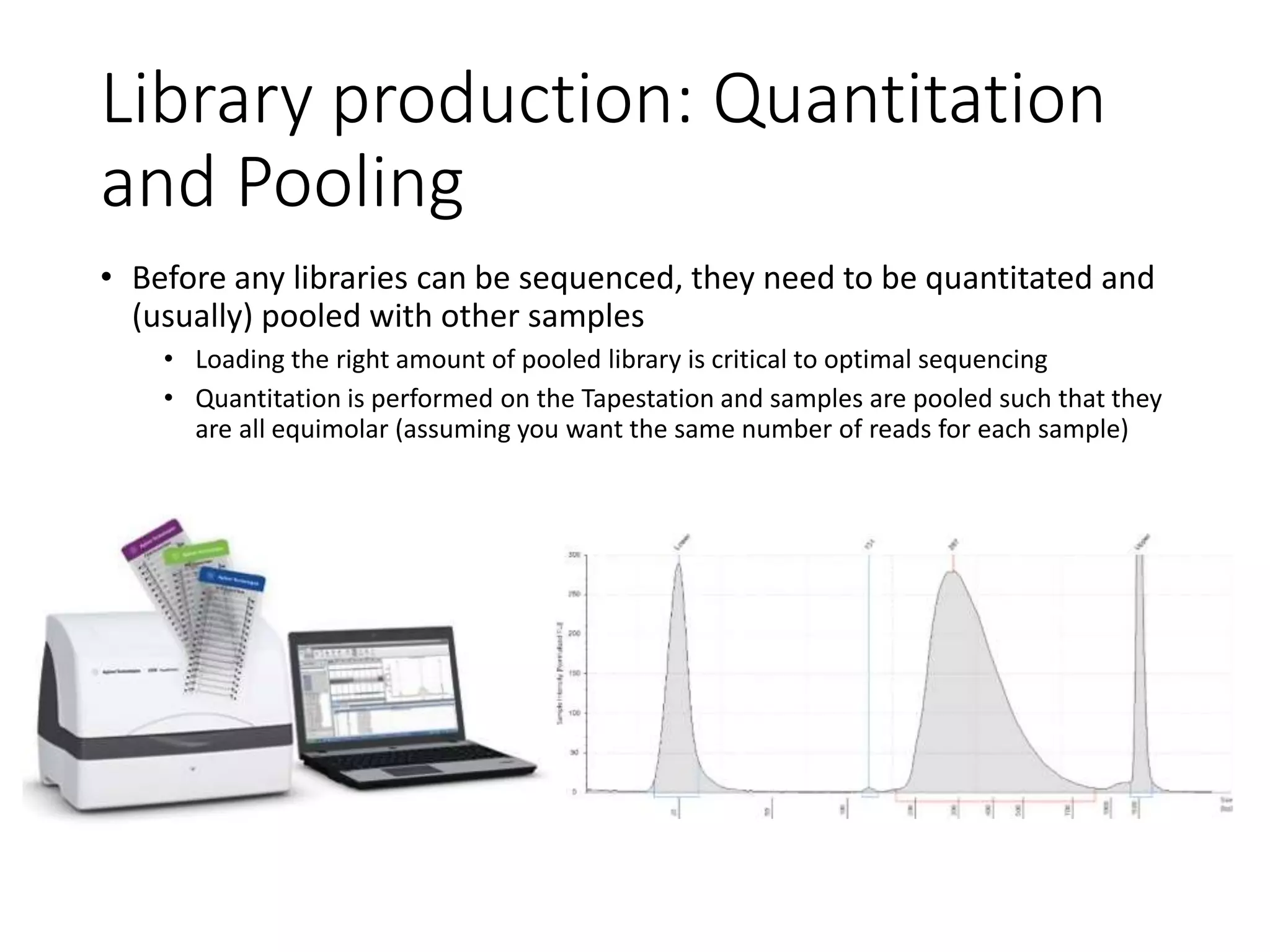
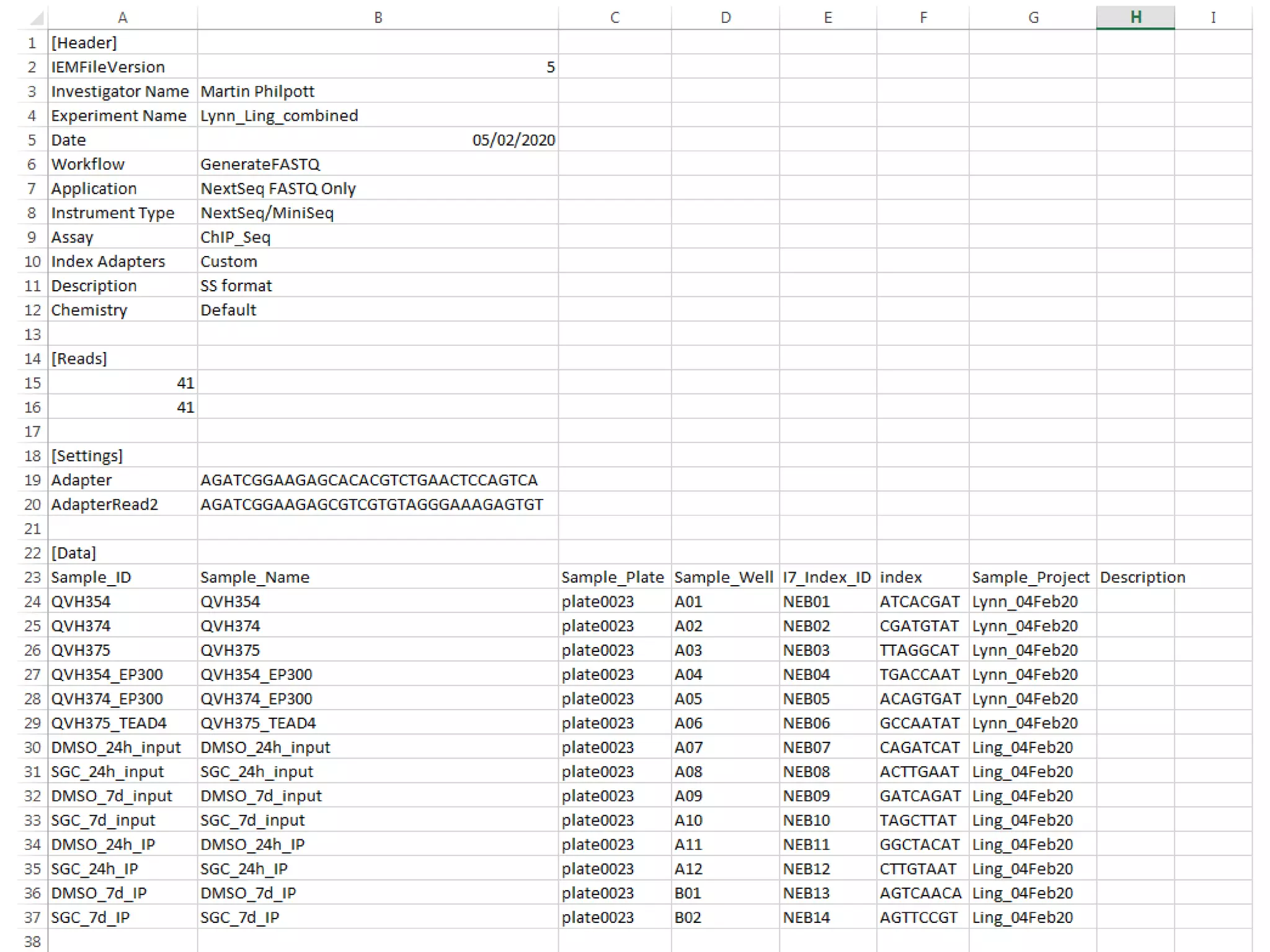
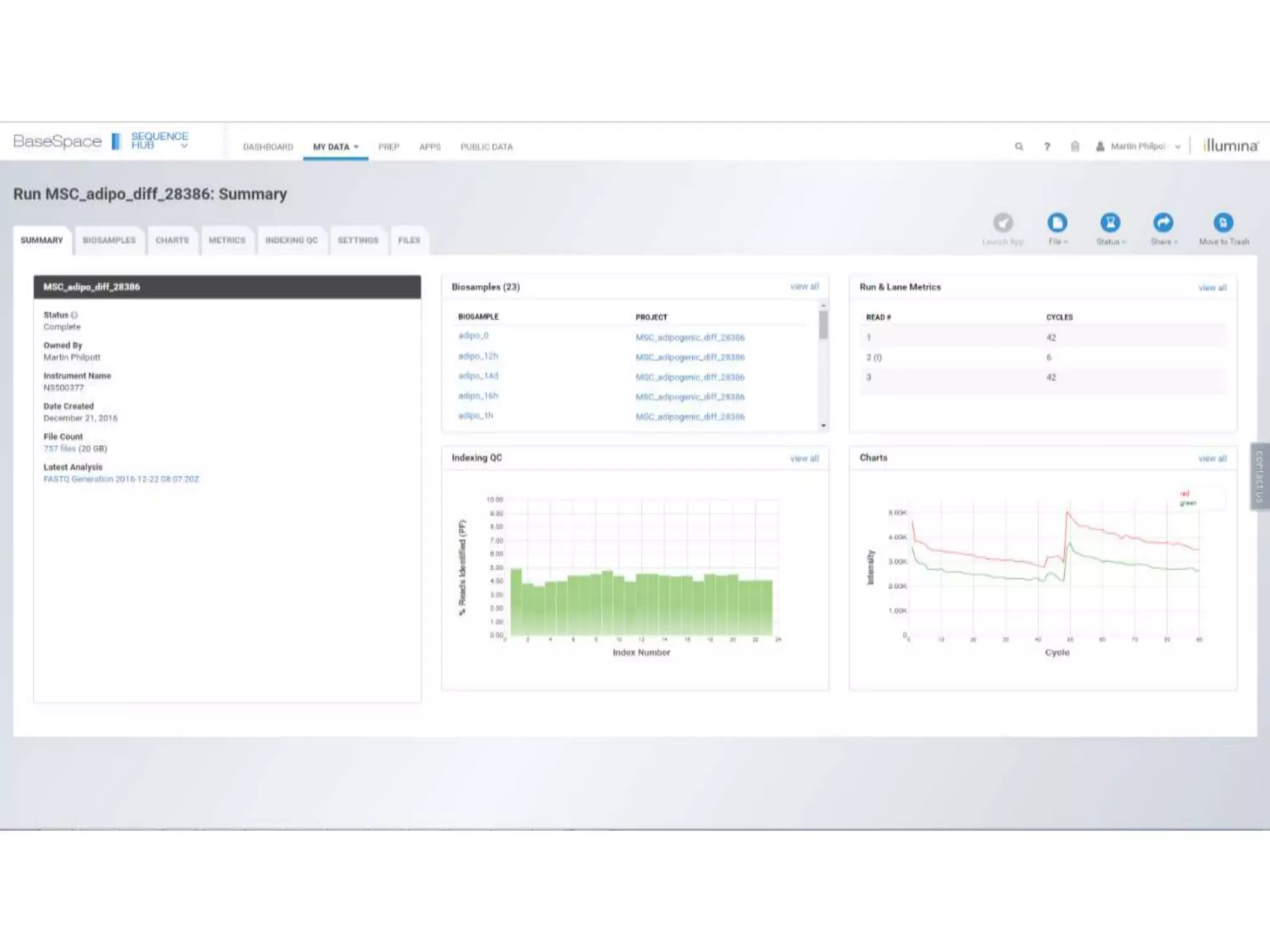
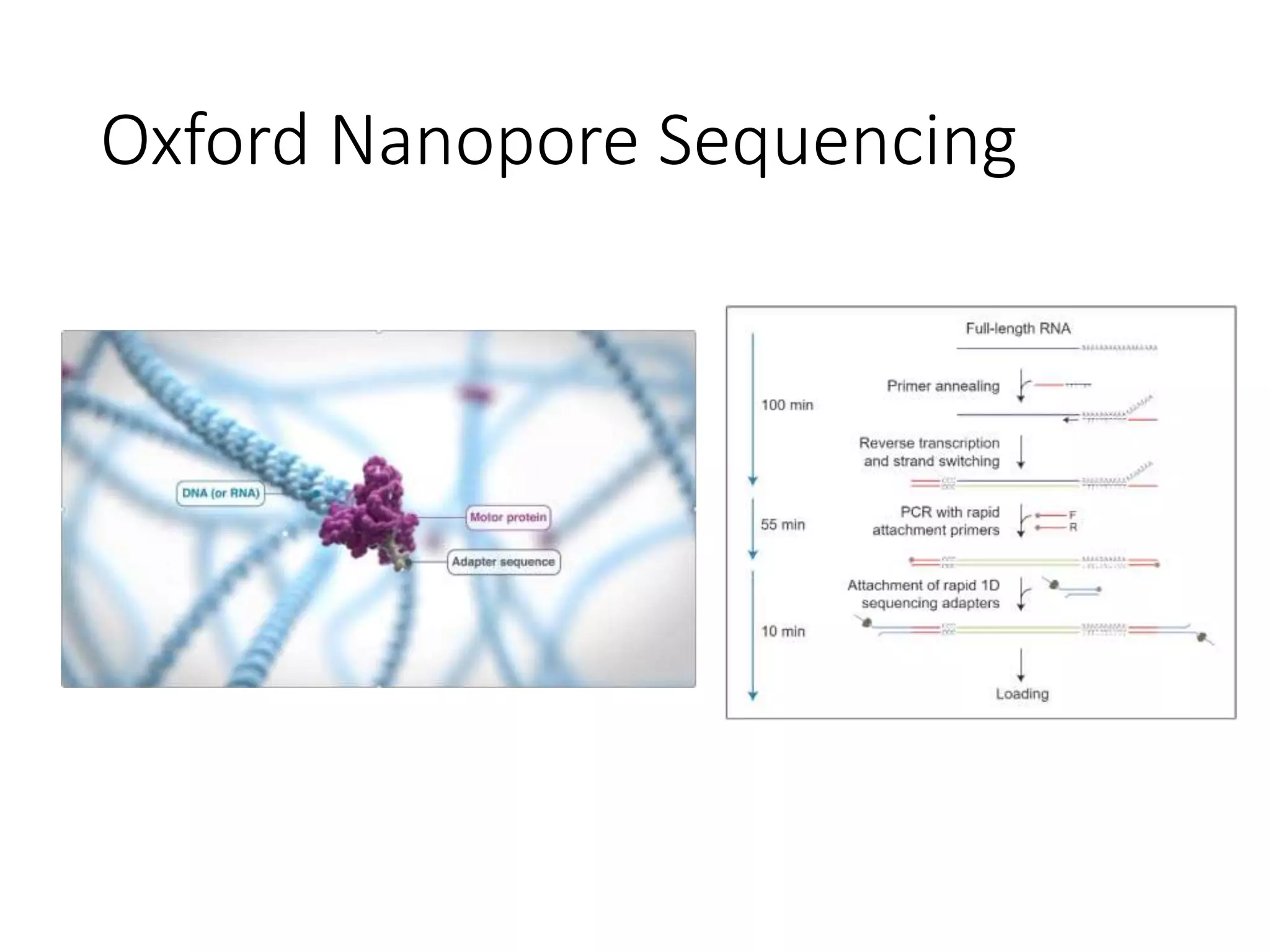
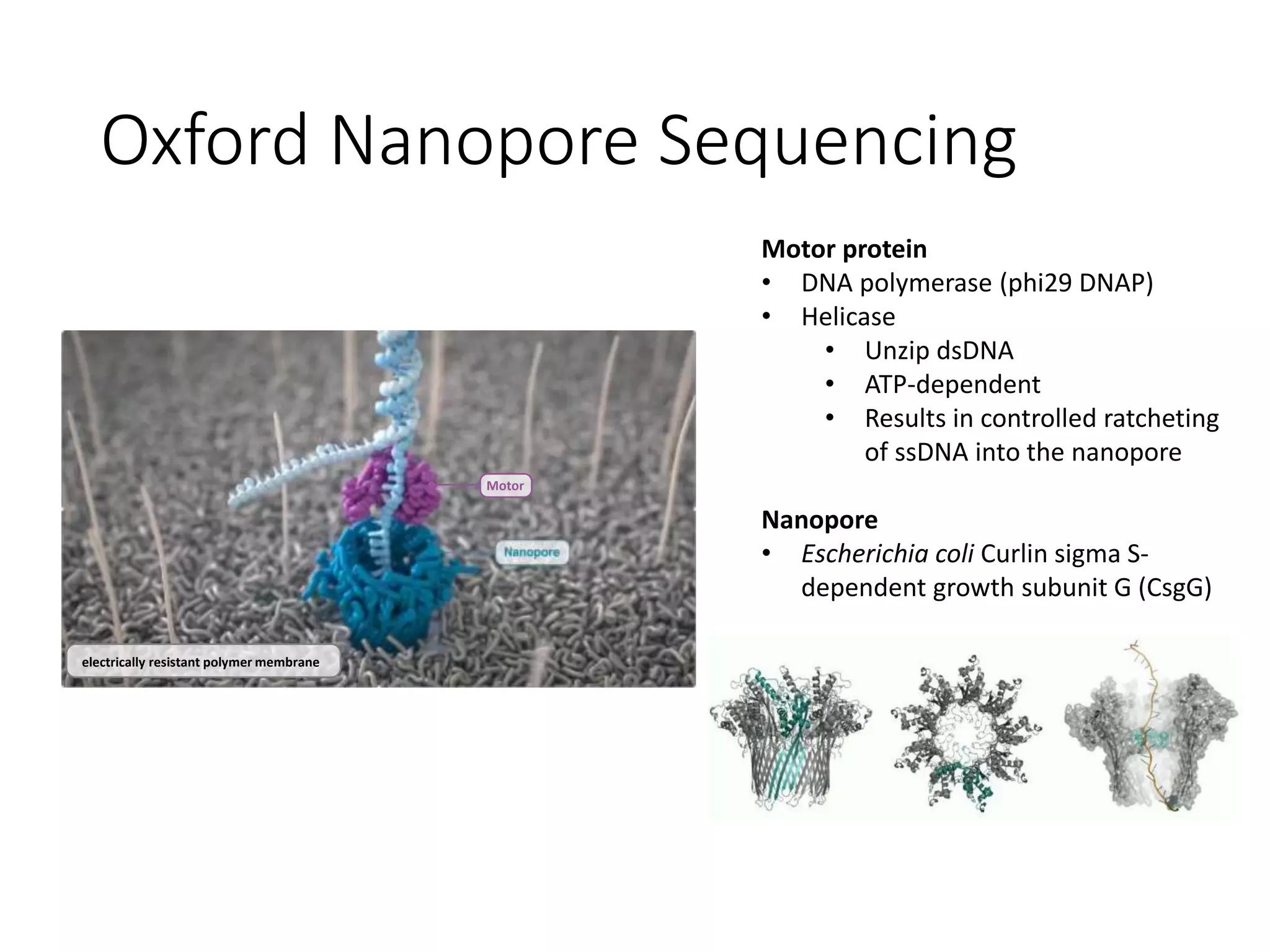
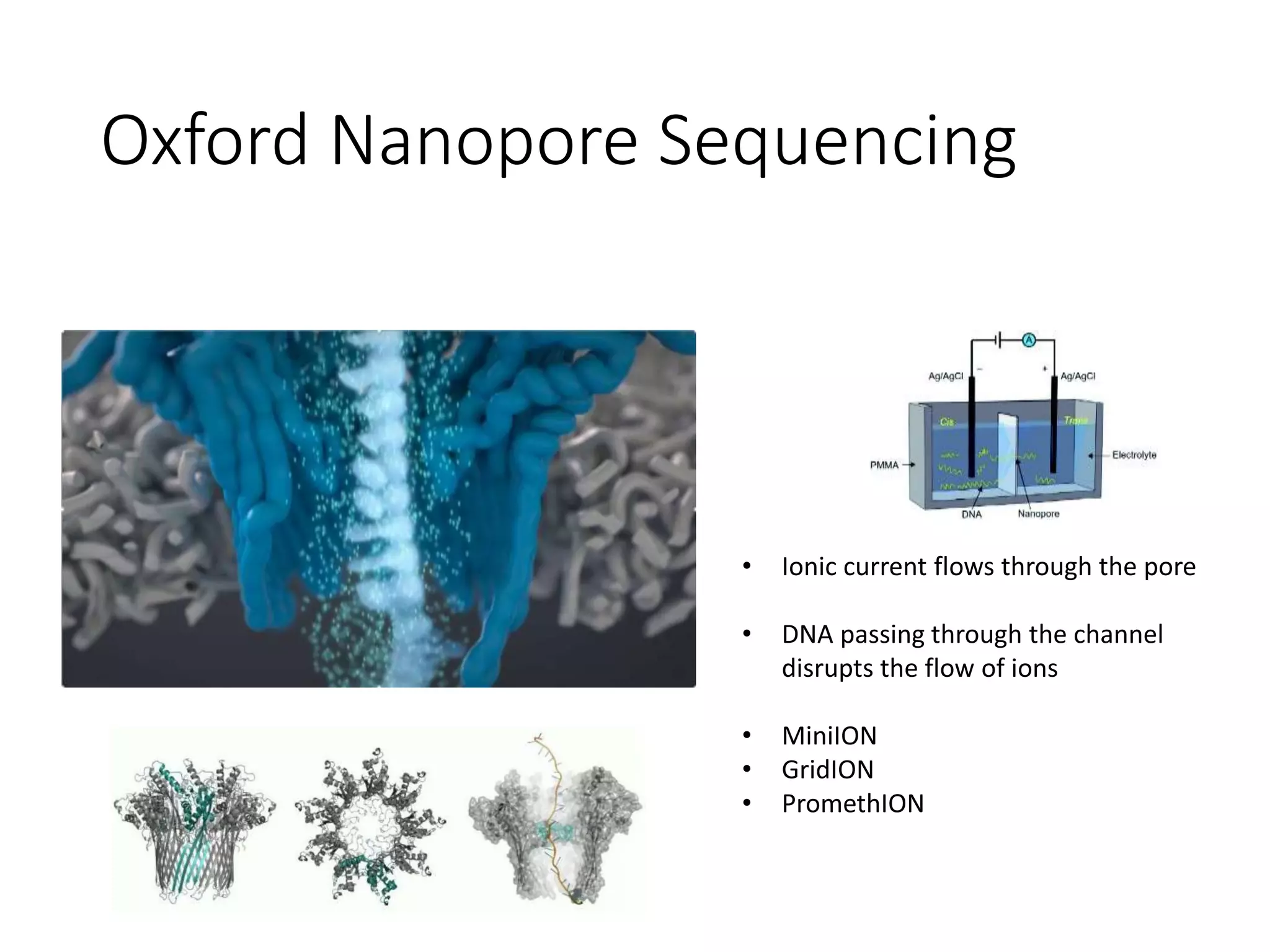
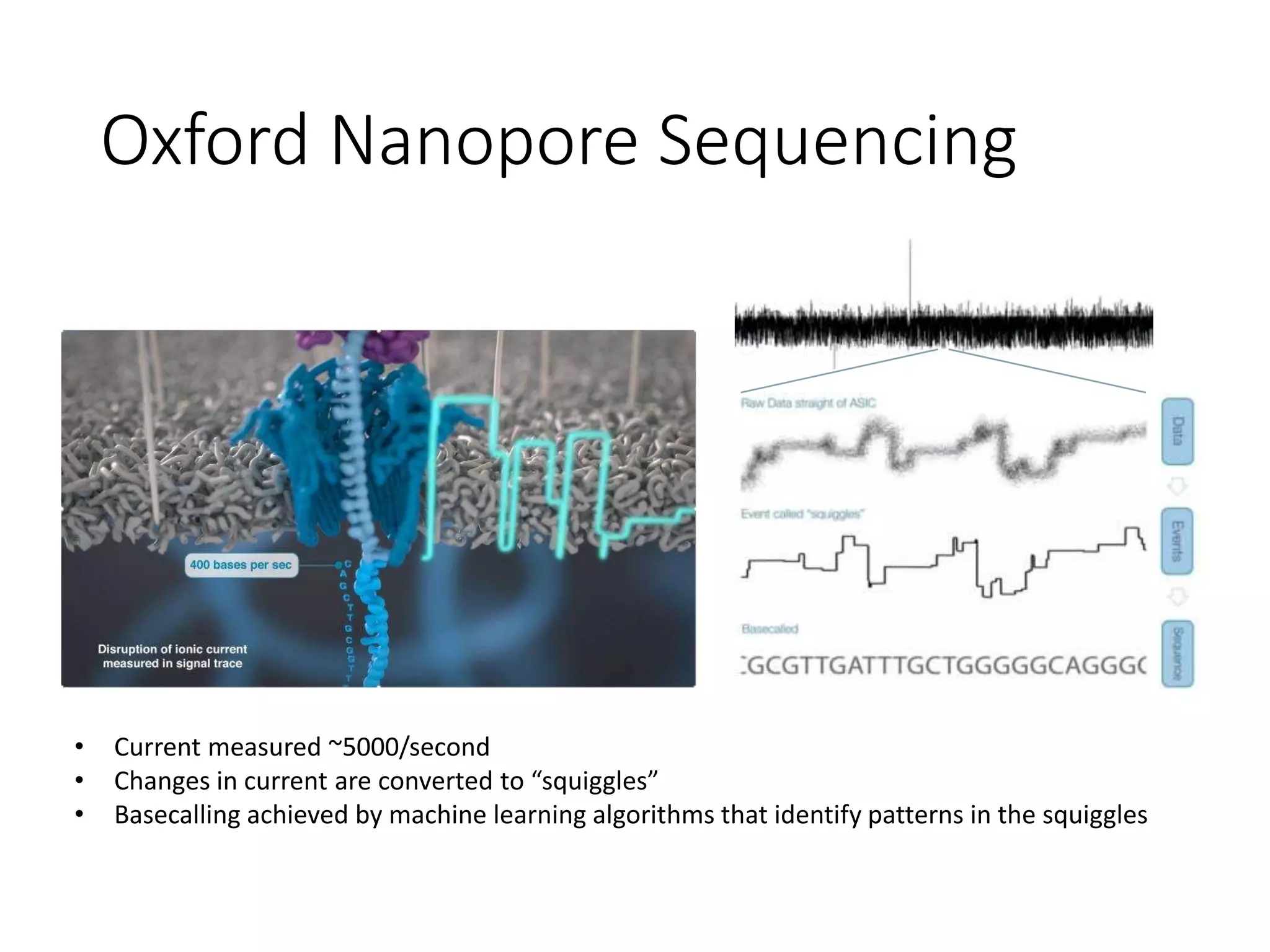
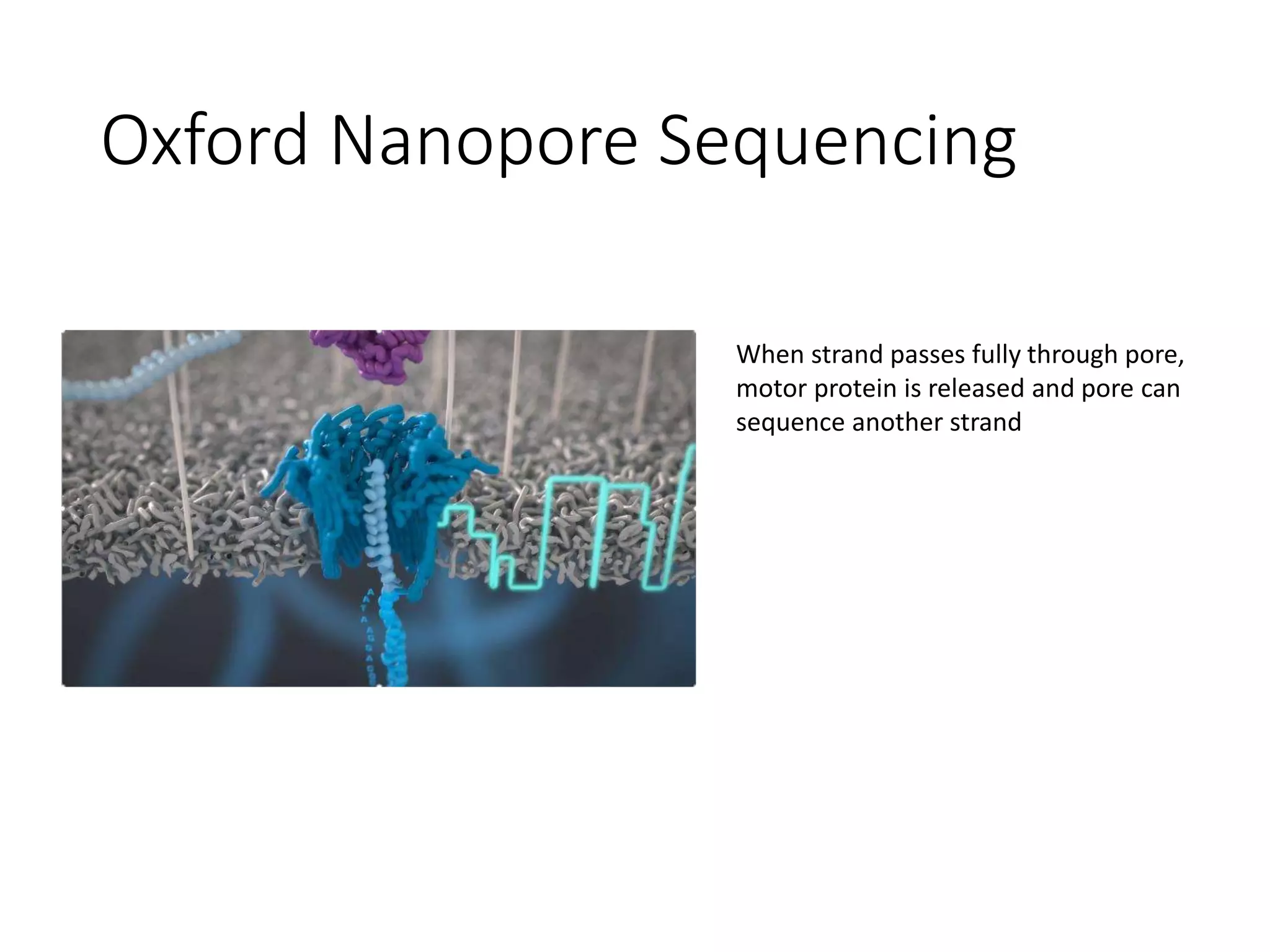
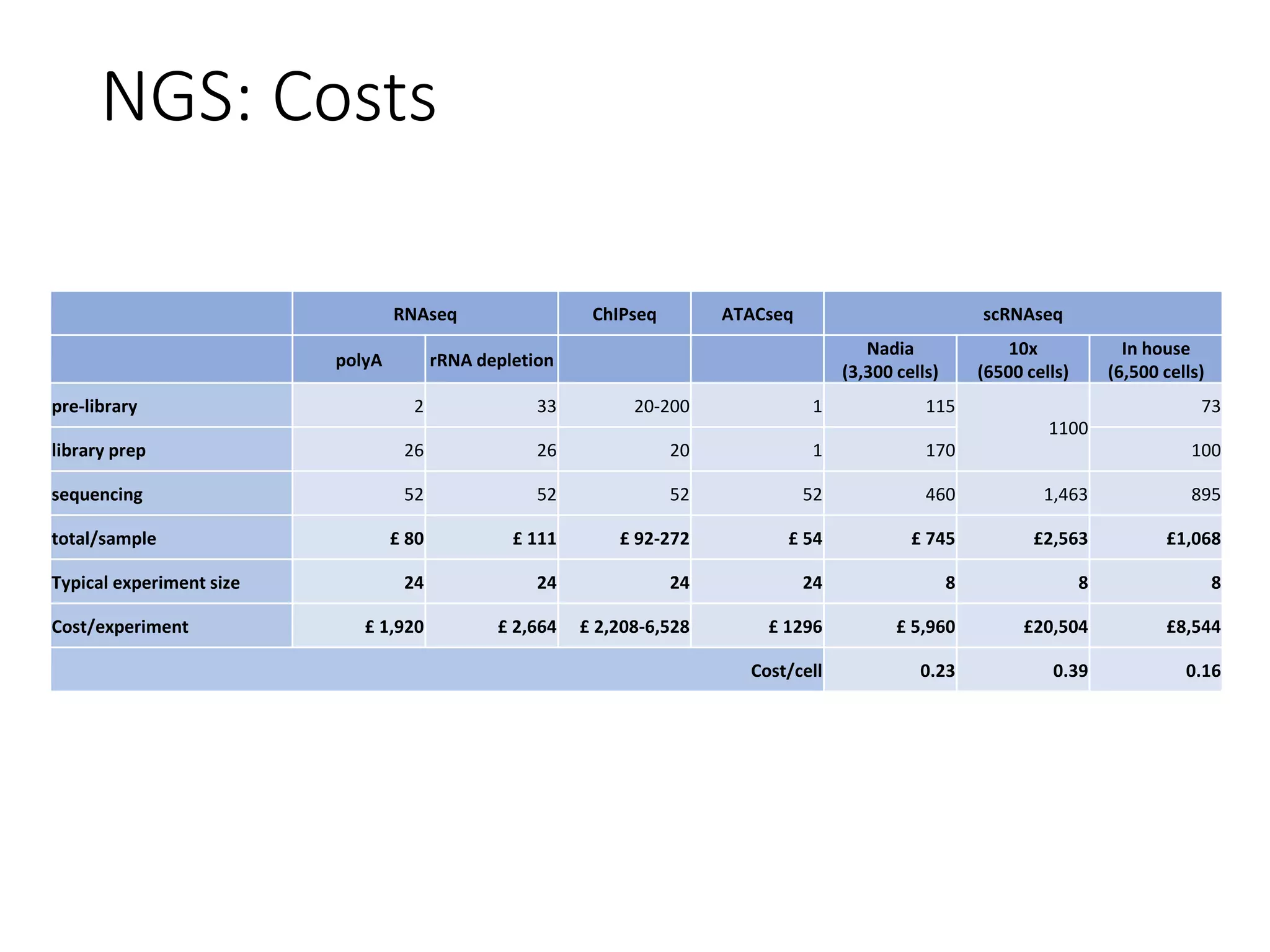
The document provides an extensive overview of next-generation sequencing (NGS) methodologies, comparing various techniques such as Sanger sequencing, pyrosequencing, Illumina sequencing, and nanopore sequencing in terms of read length, accuracy, costs, and advantages/disadvantages. It also addresses library preparation methods for RNAseq, ChIPseq, and ATACseq, detailing specific protocols and considerations for different sequencing applications. The cost analysis of experiments and the significance of accurately quantitating and pooling libraries for sequencing are also discussed.