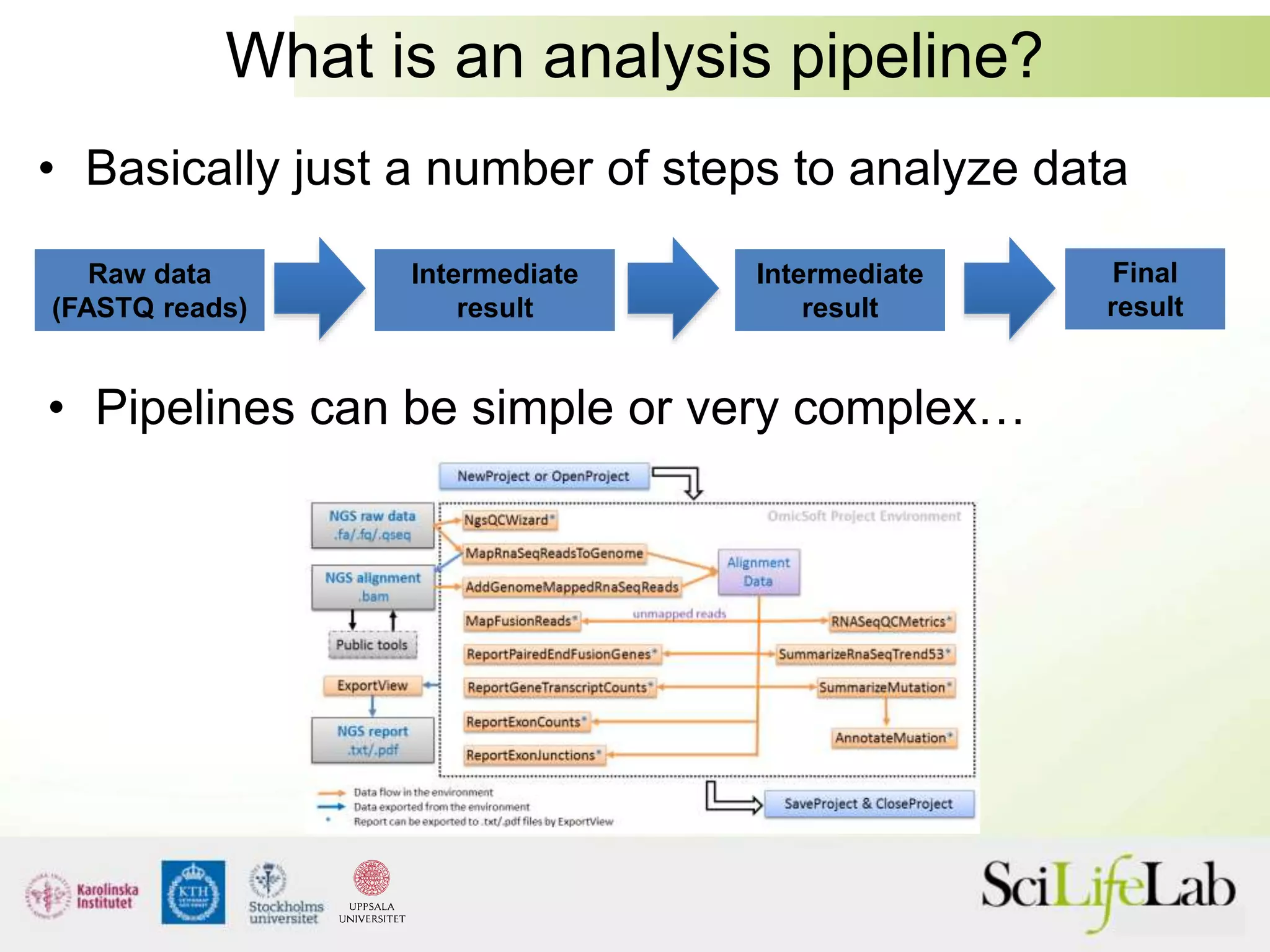
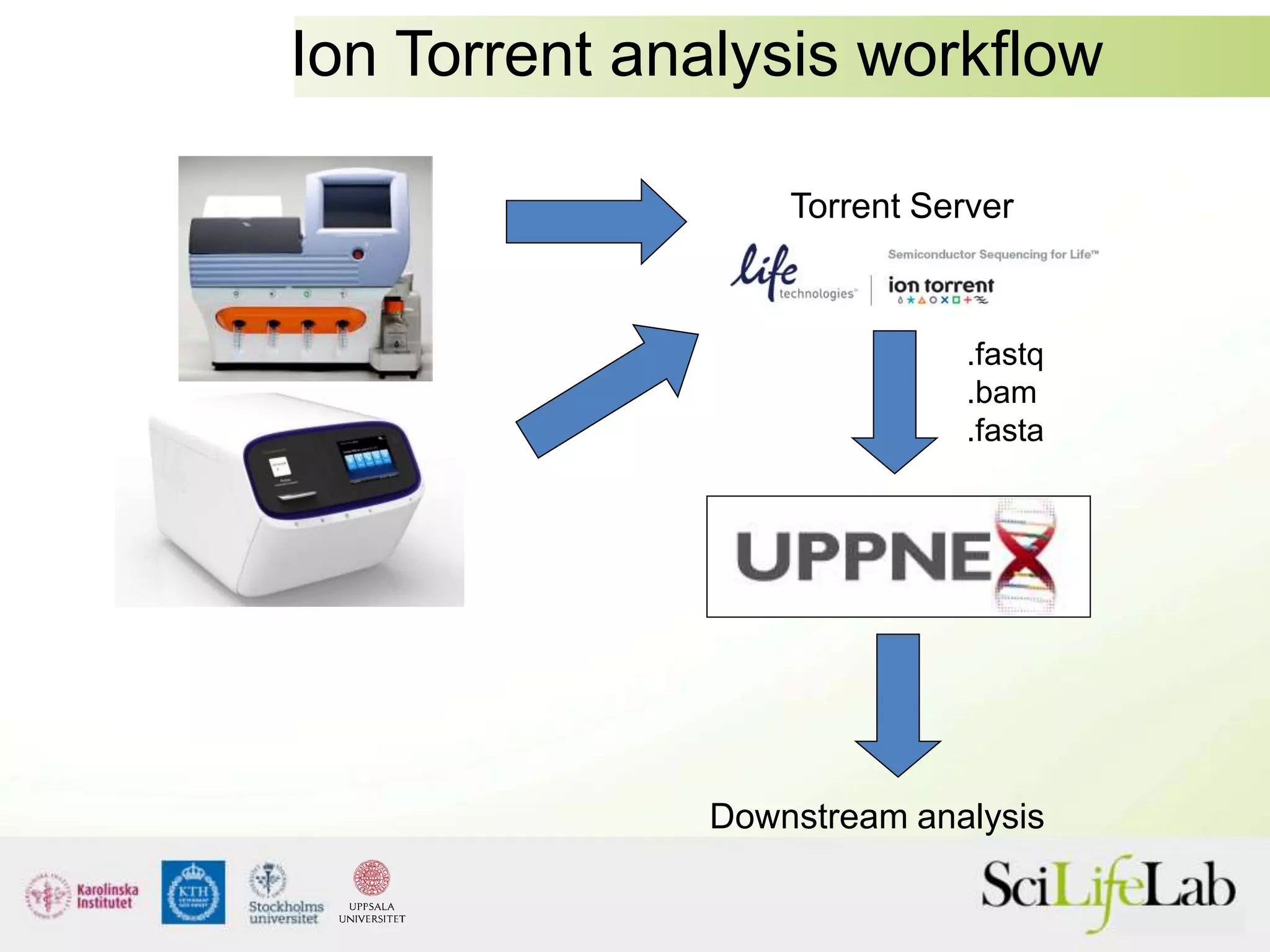
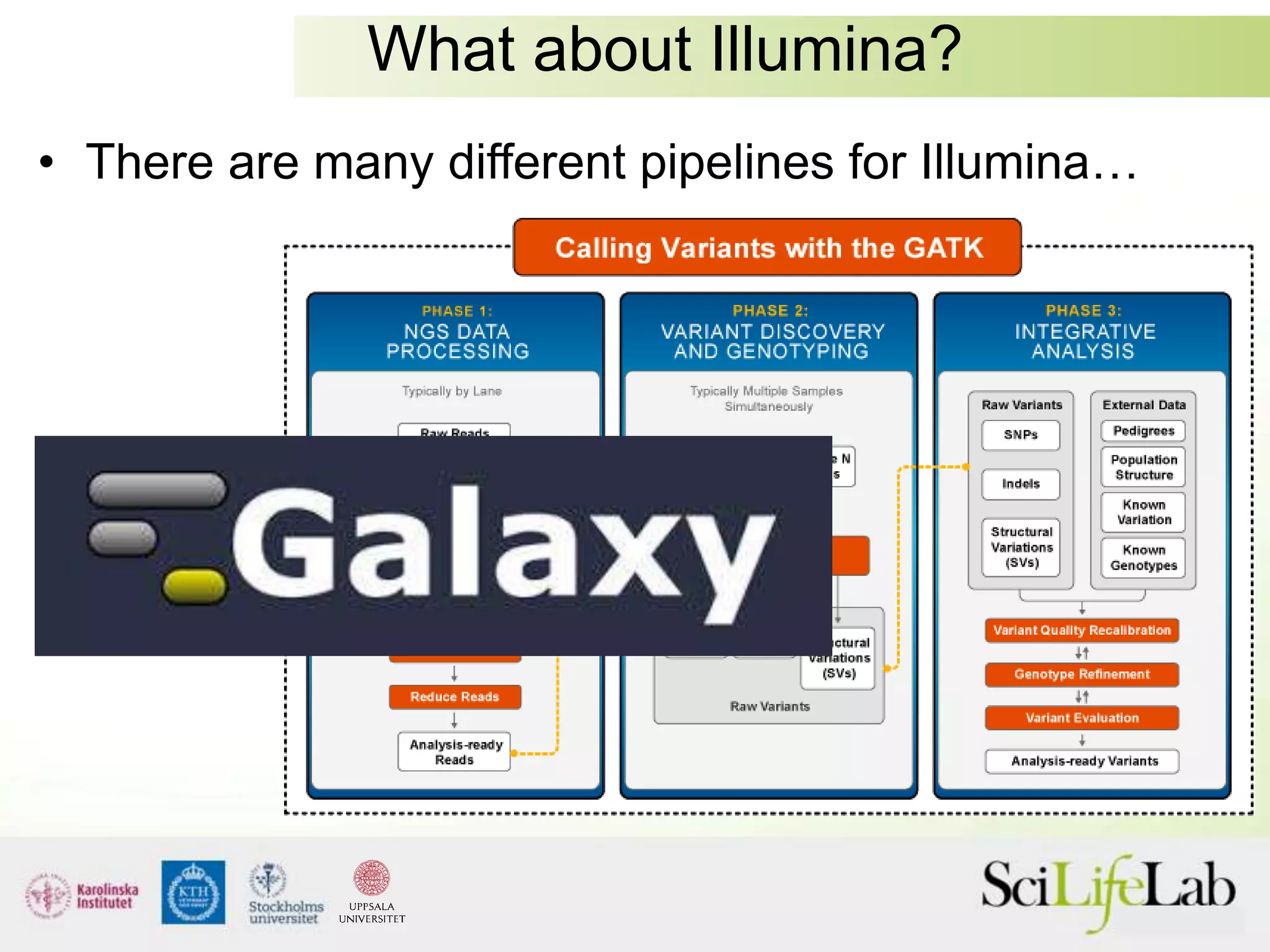
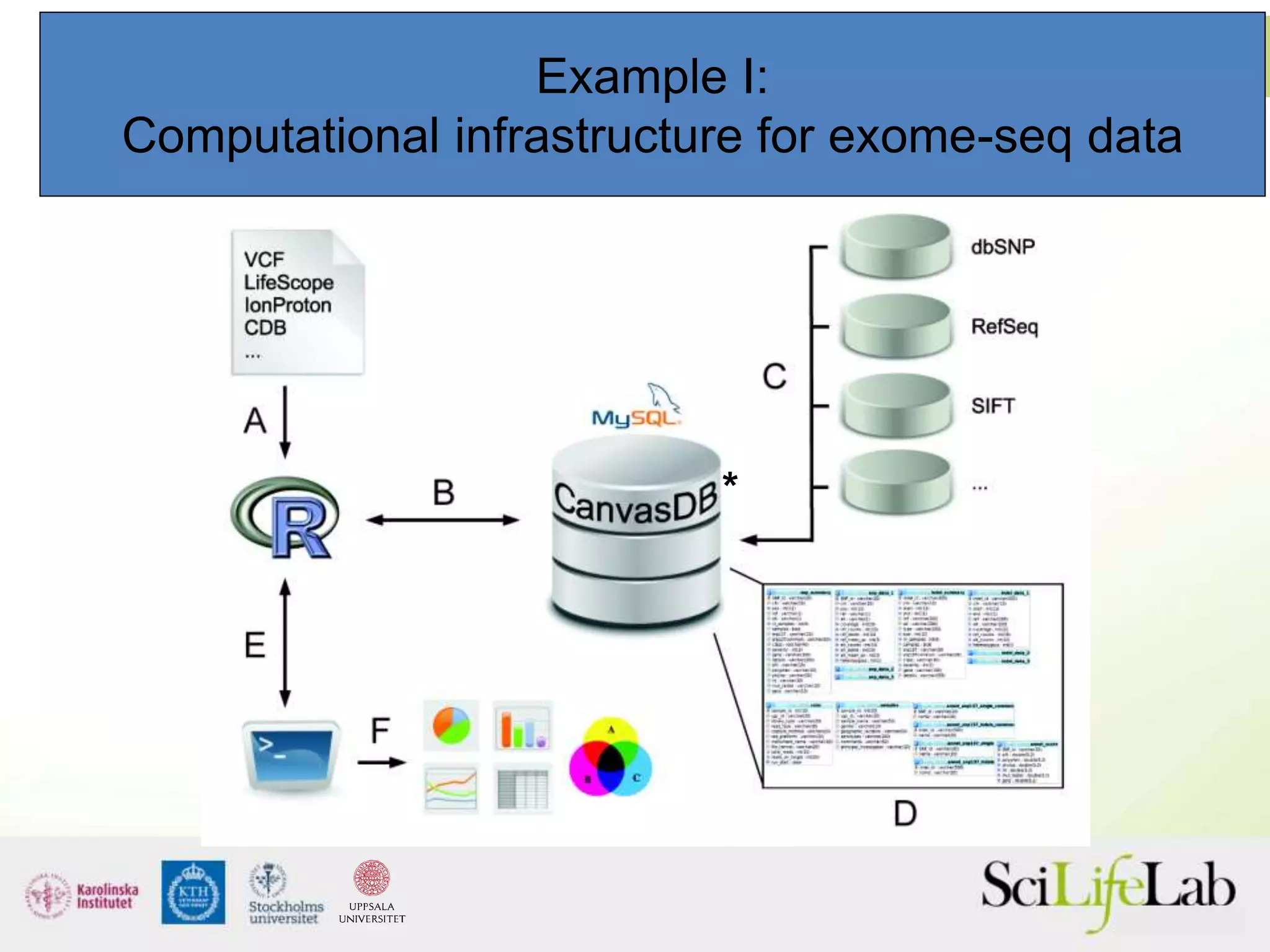
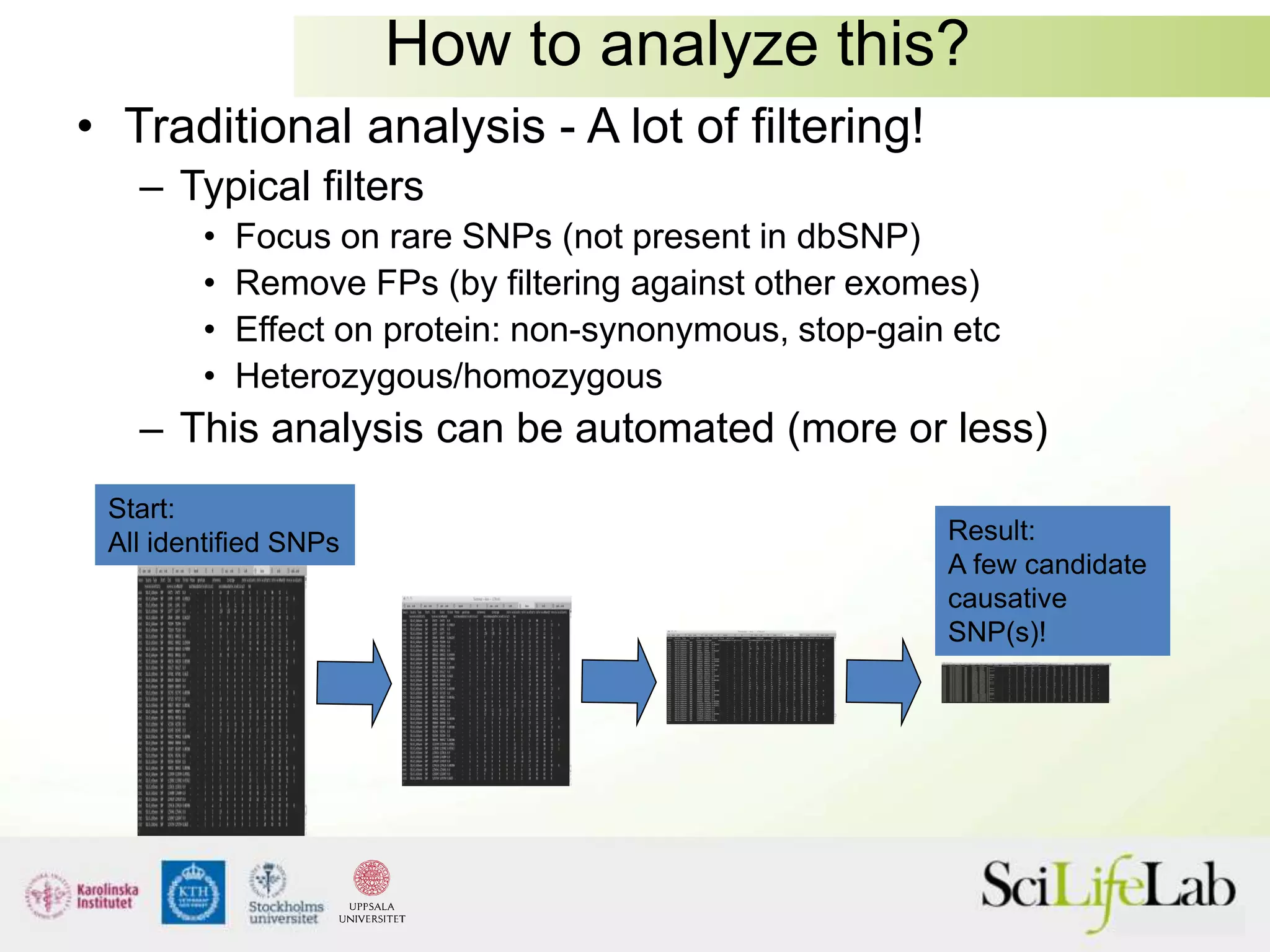
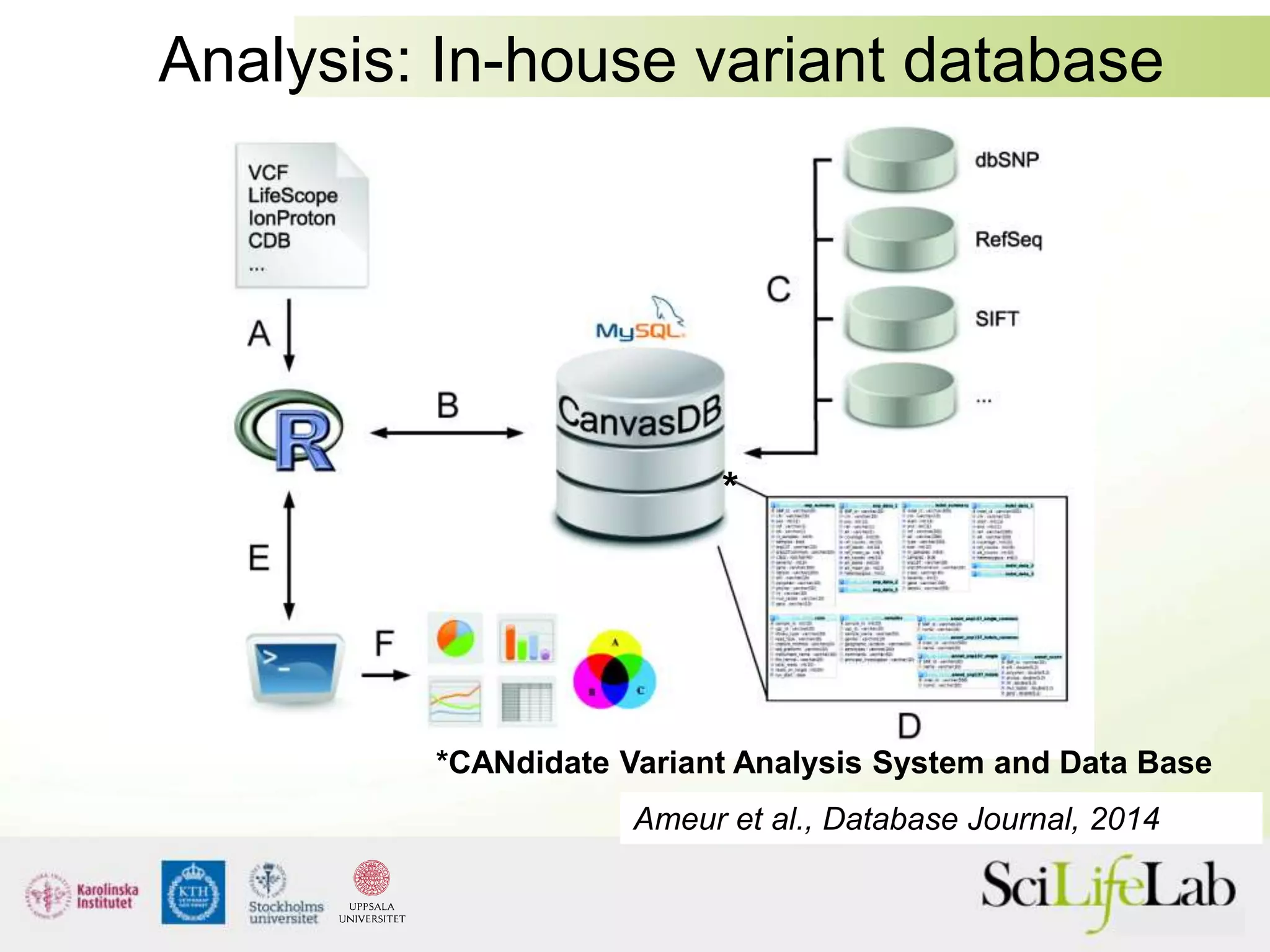
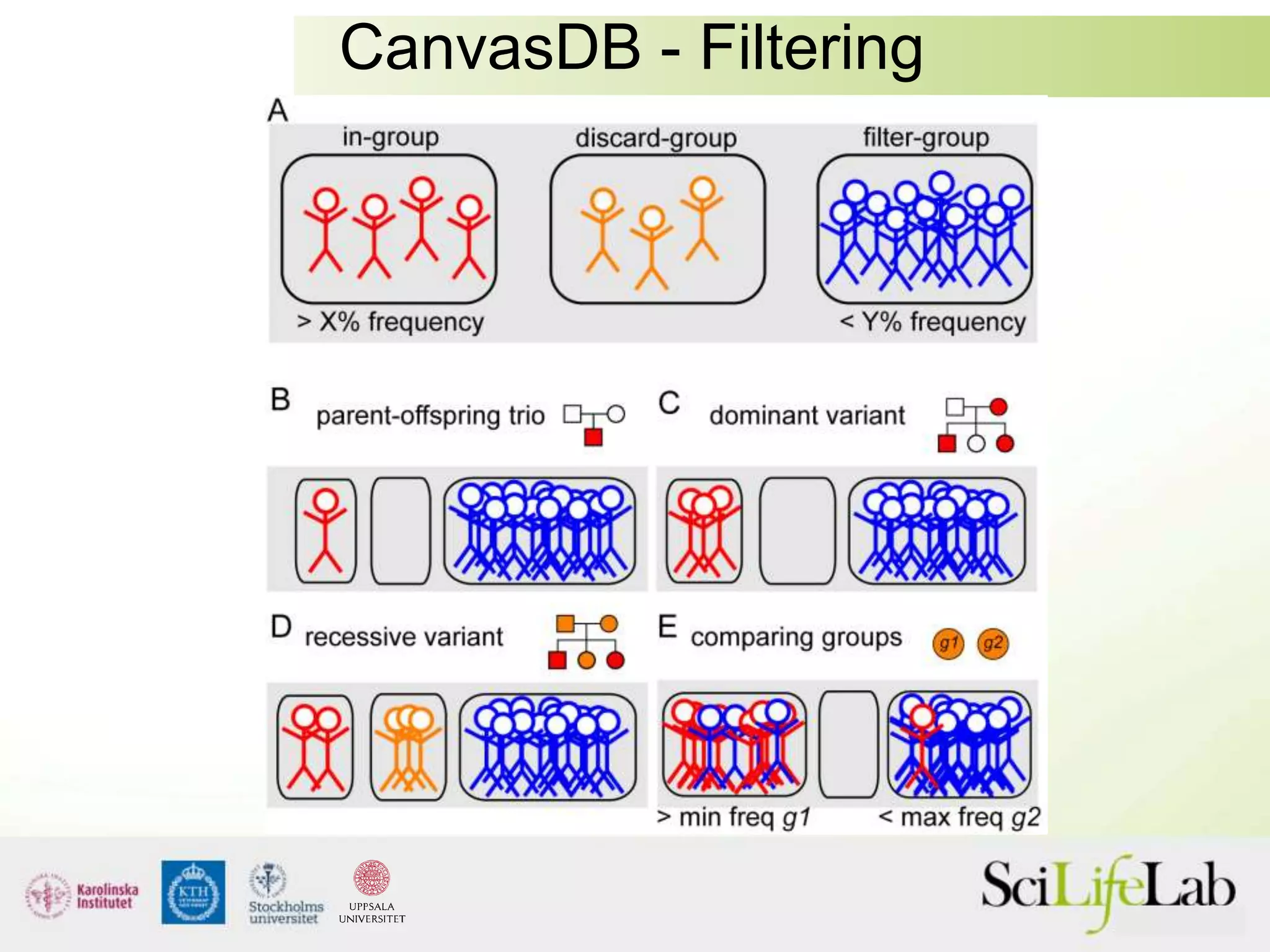
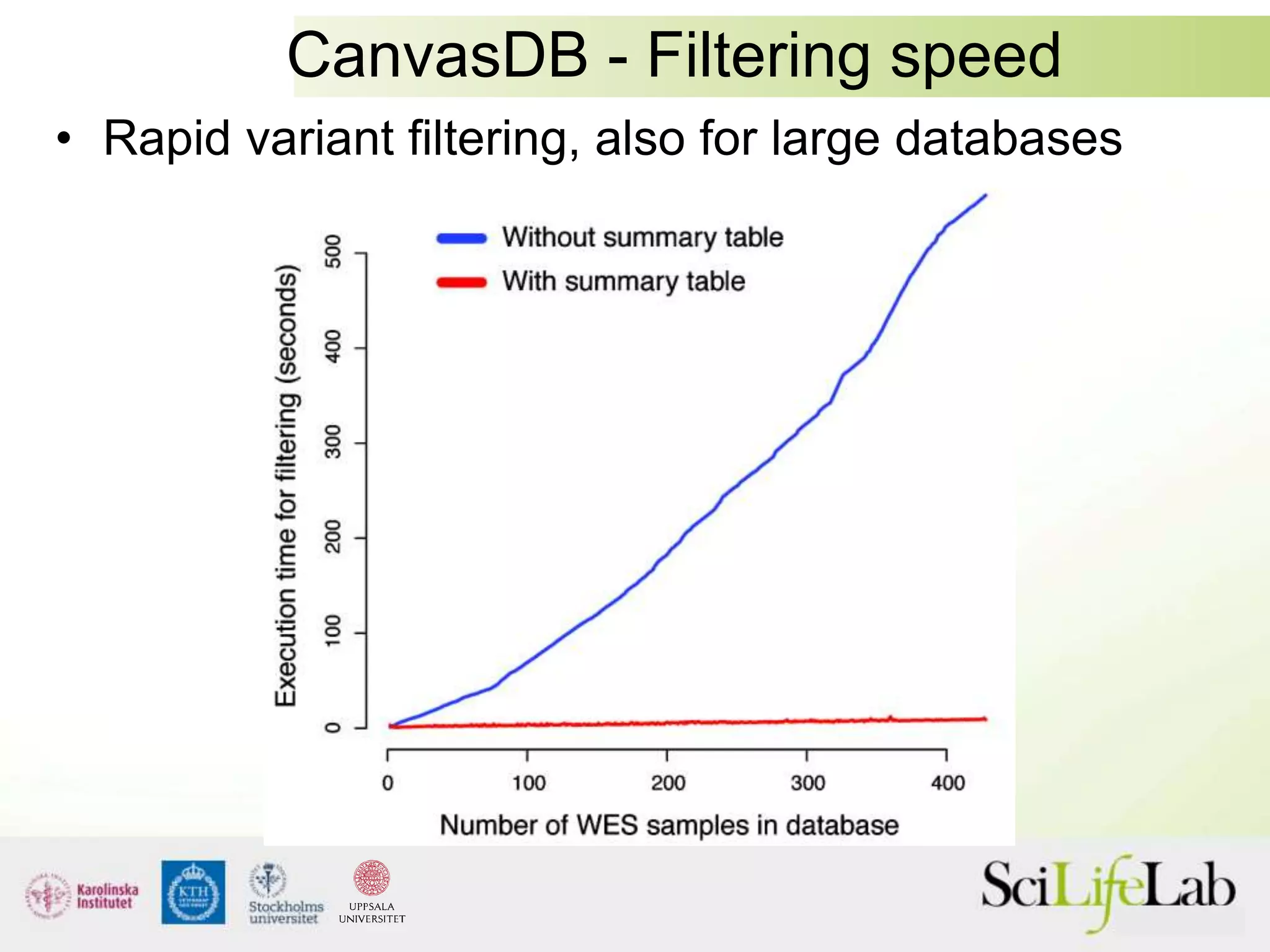
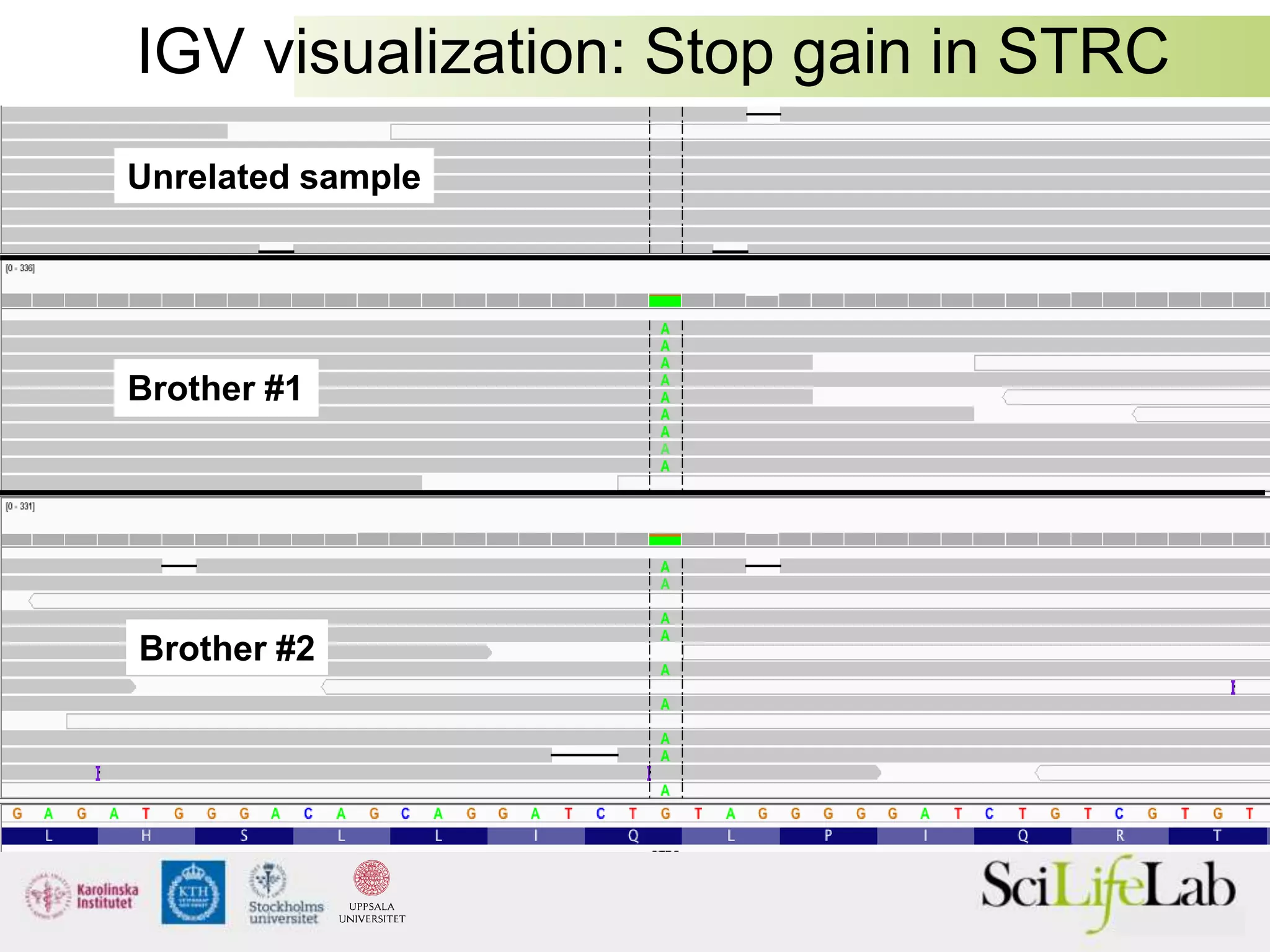
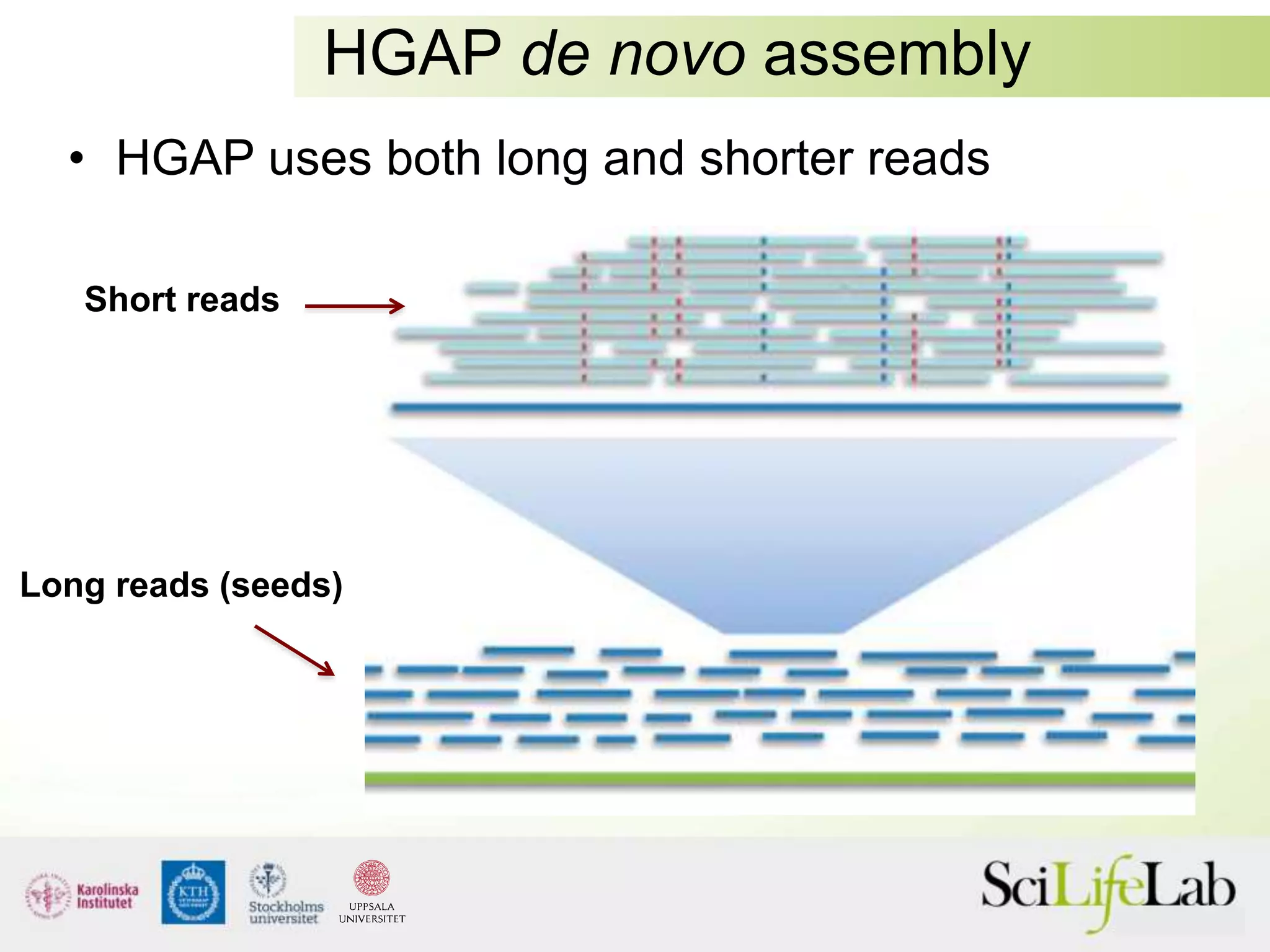
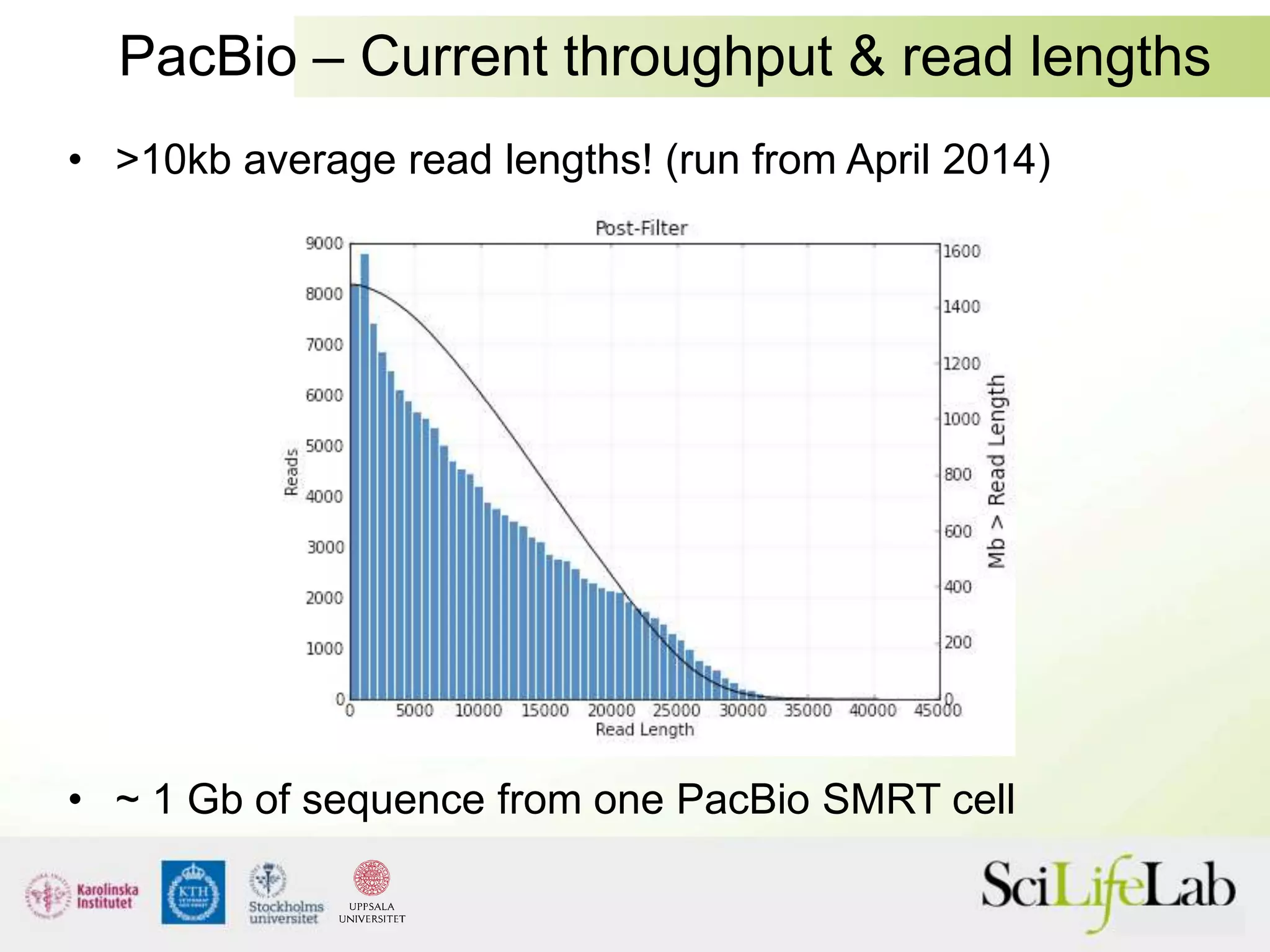
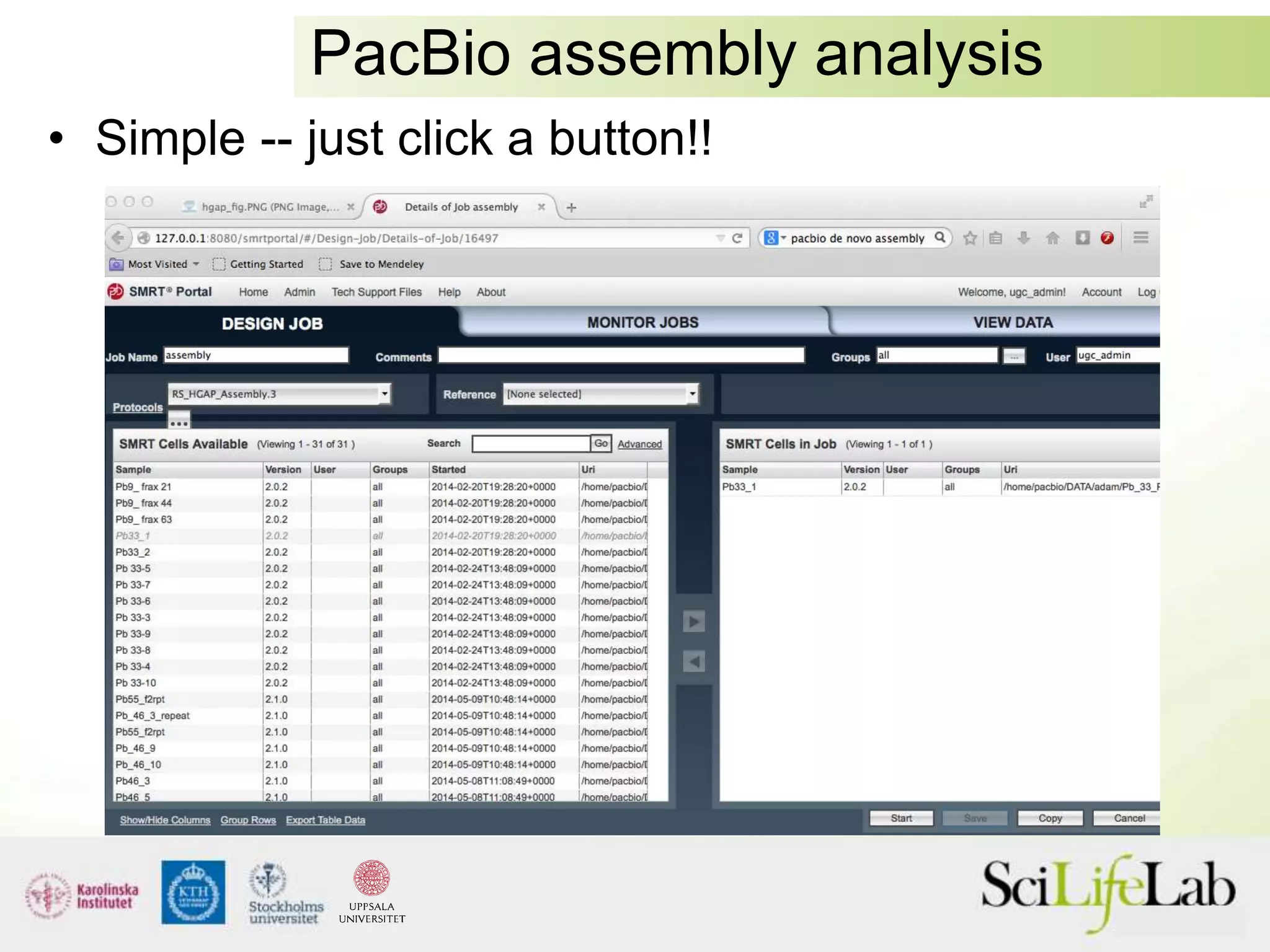
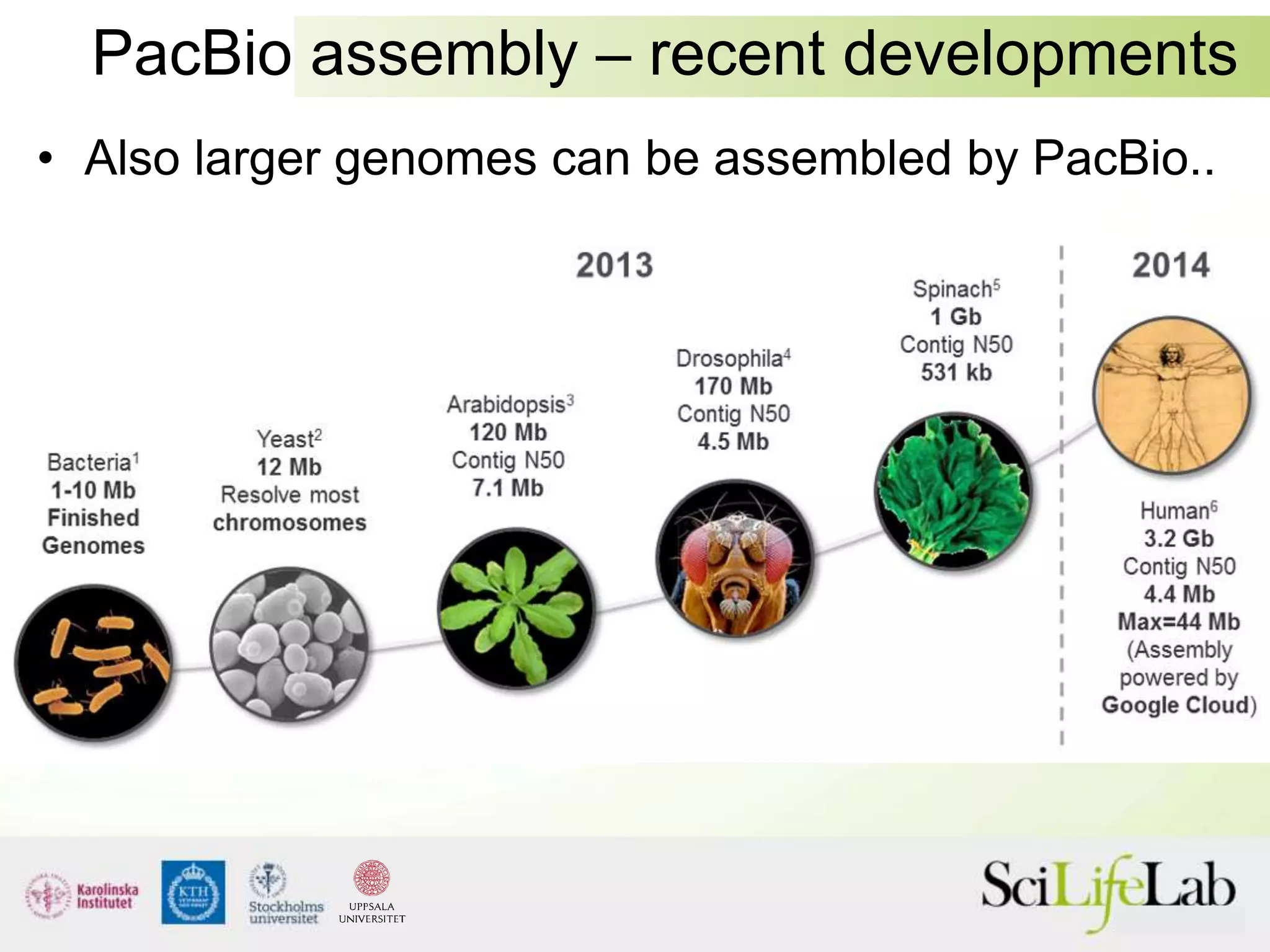
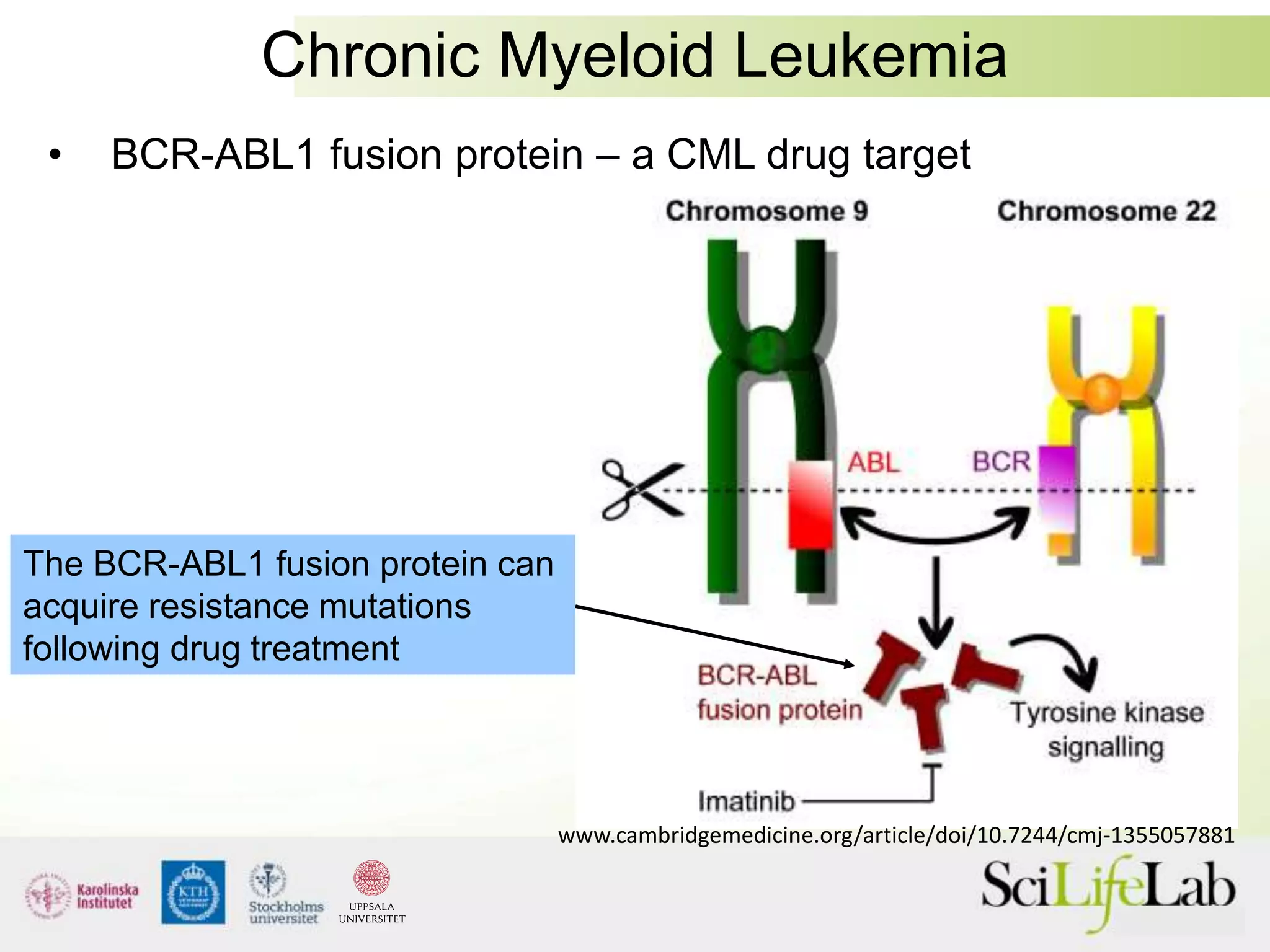
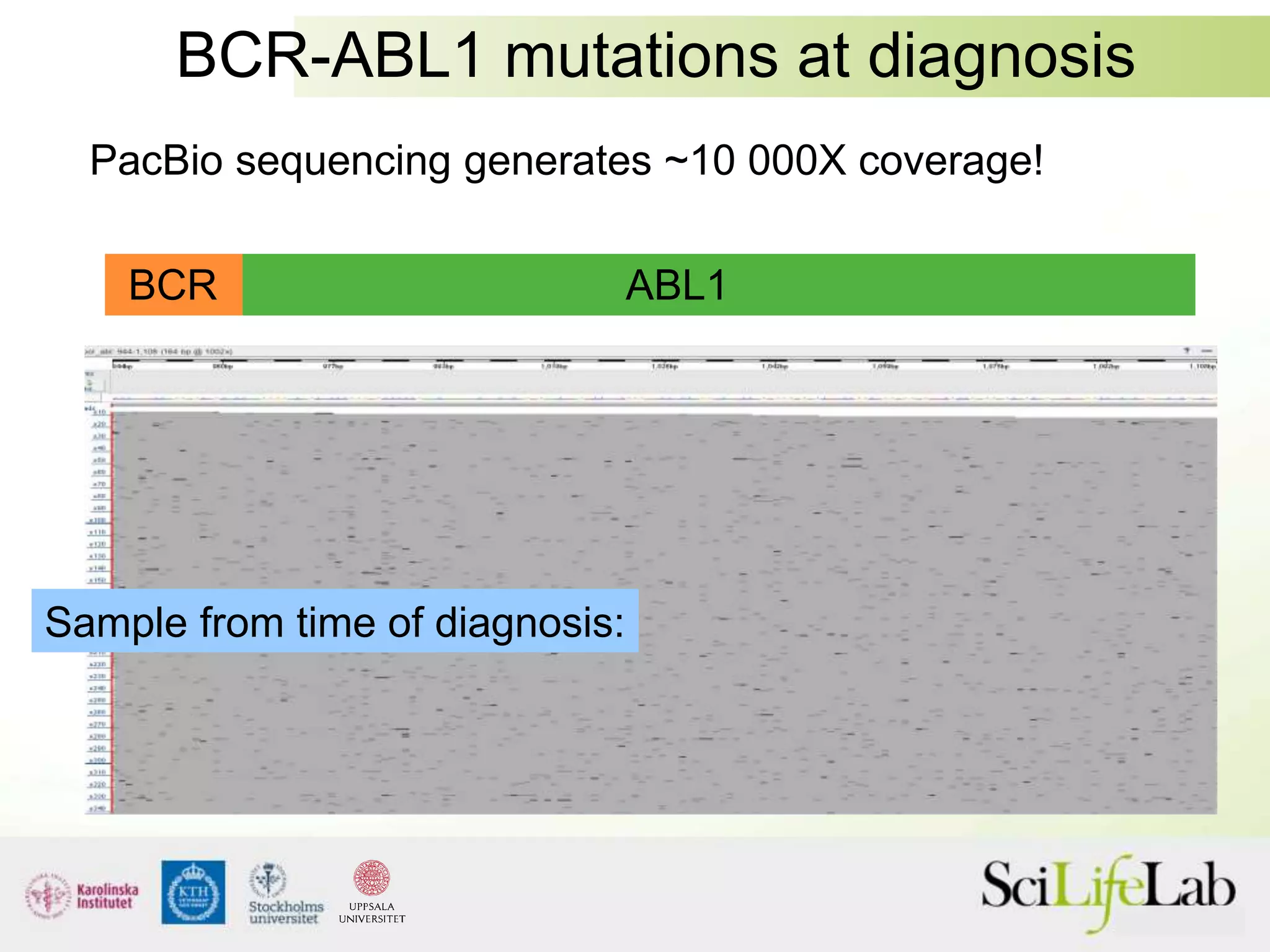
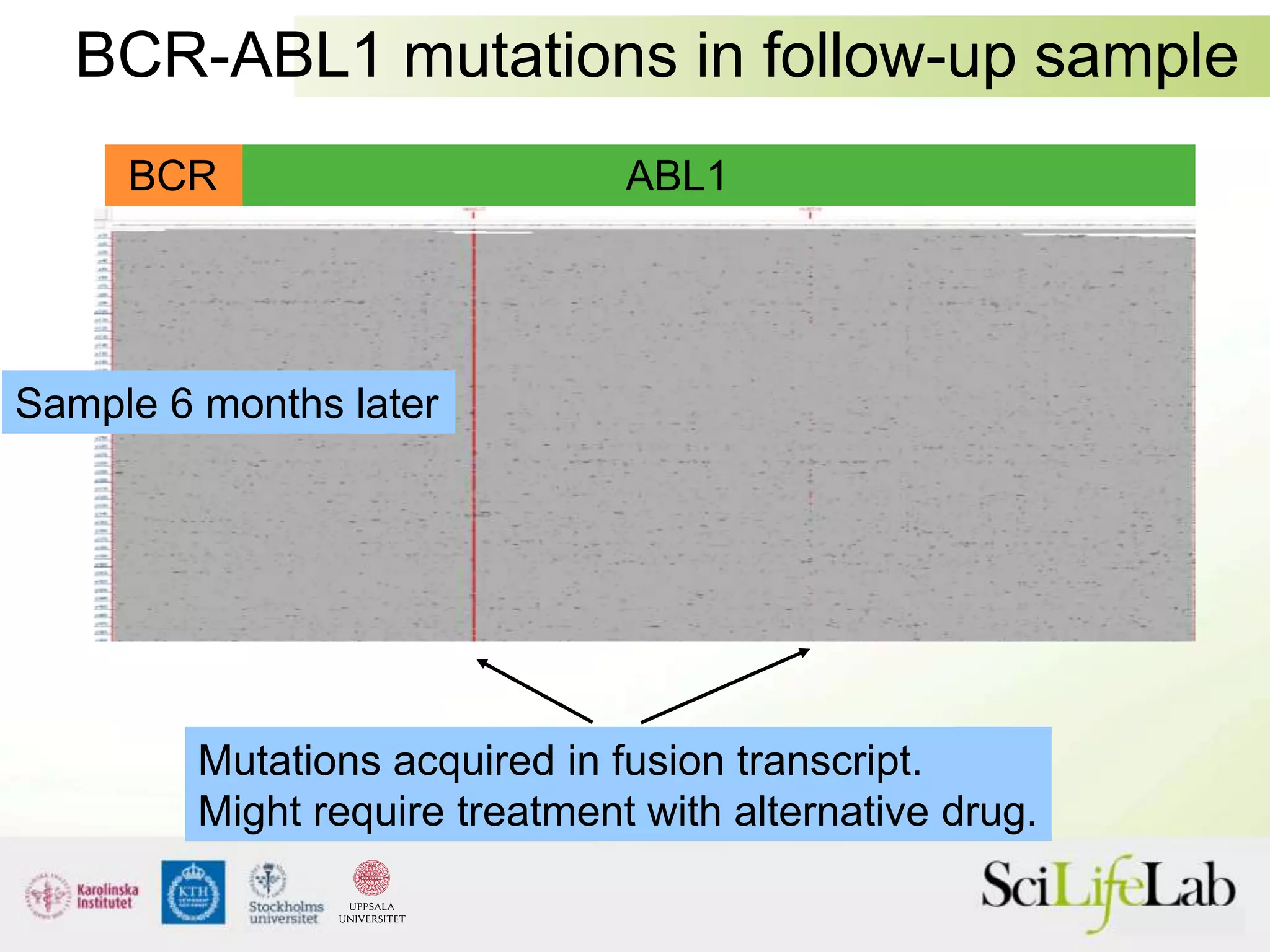
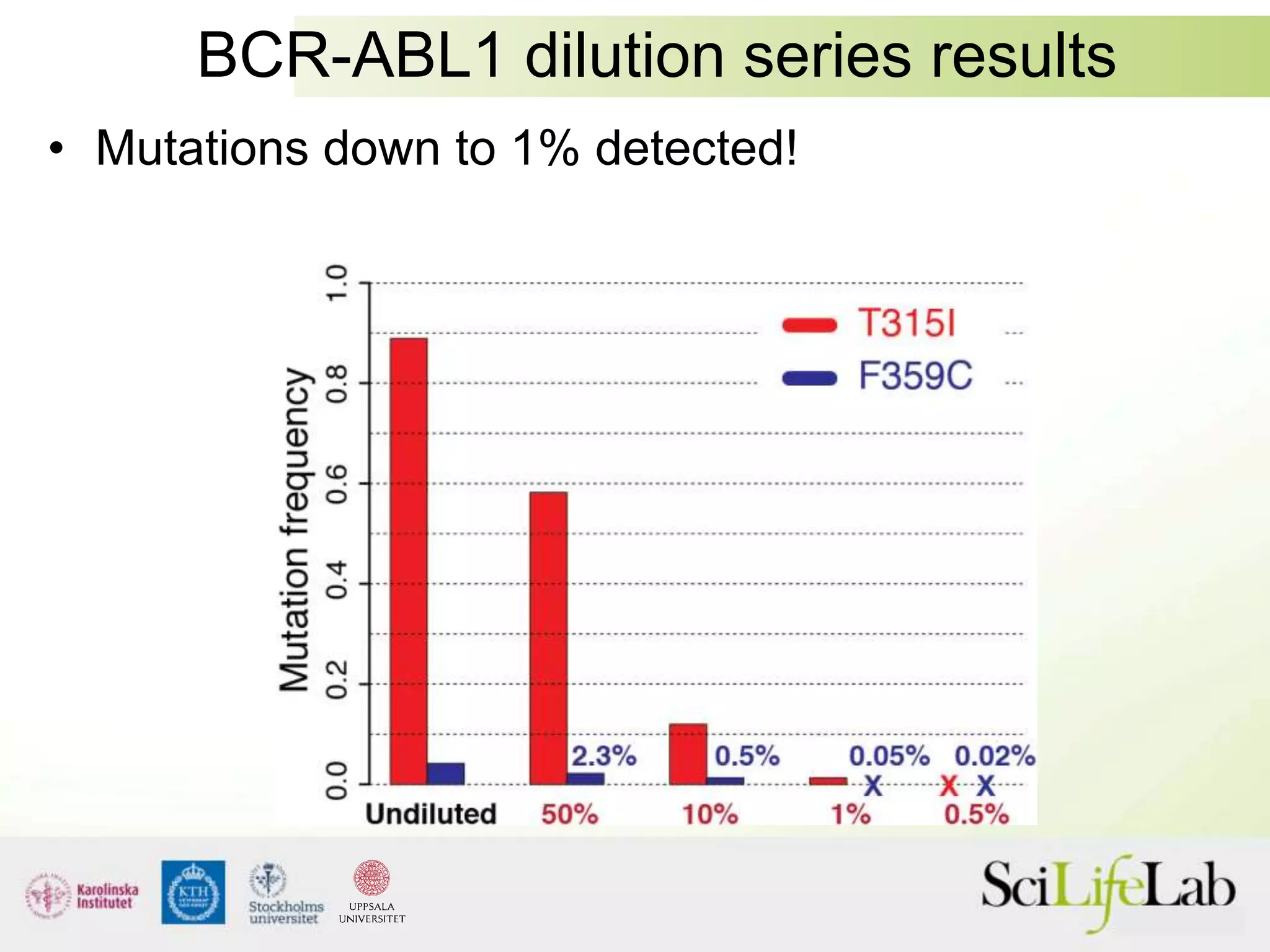
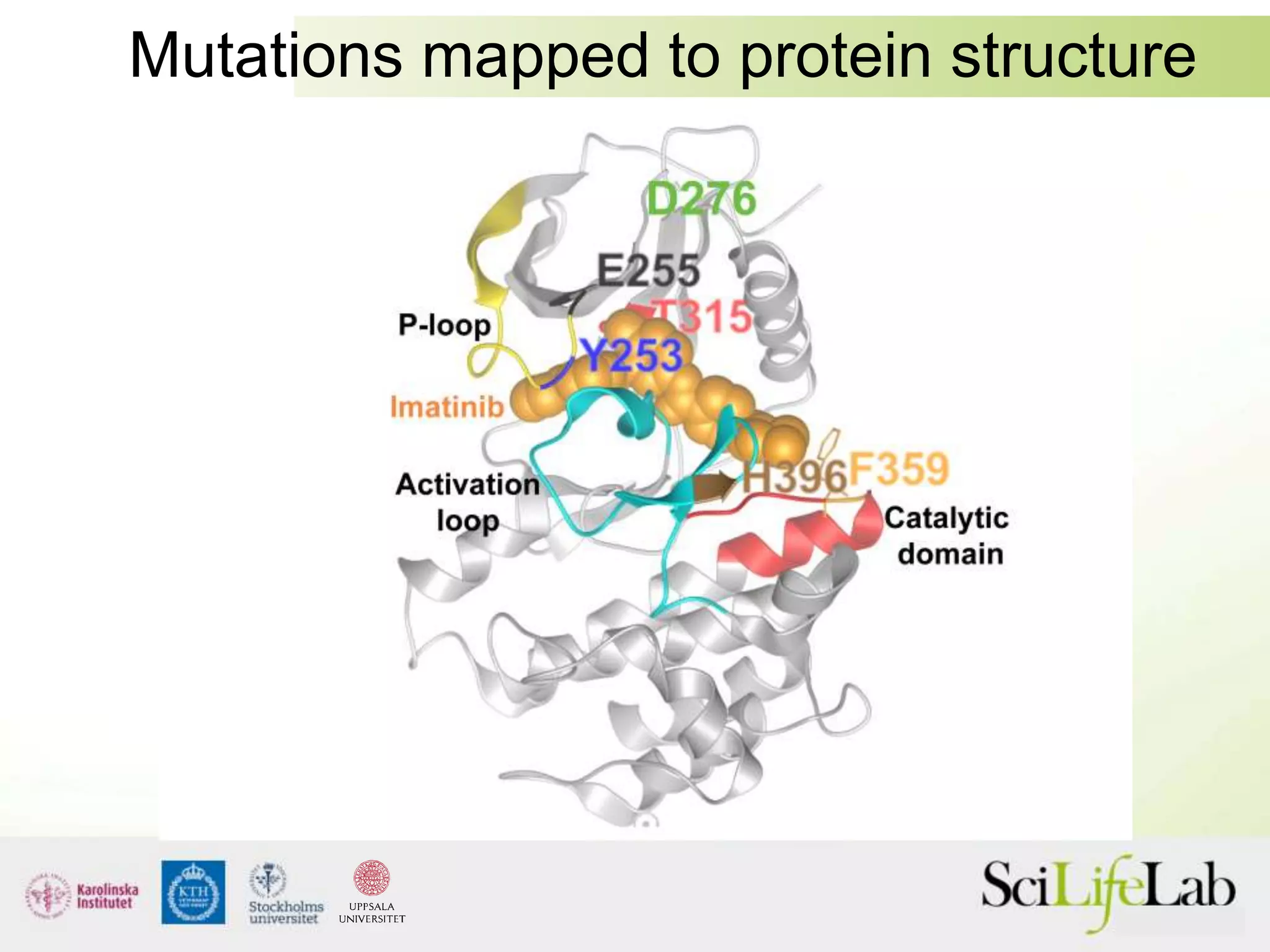
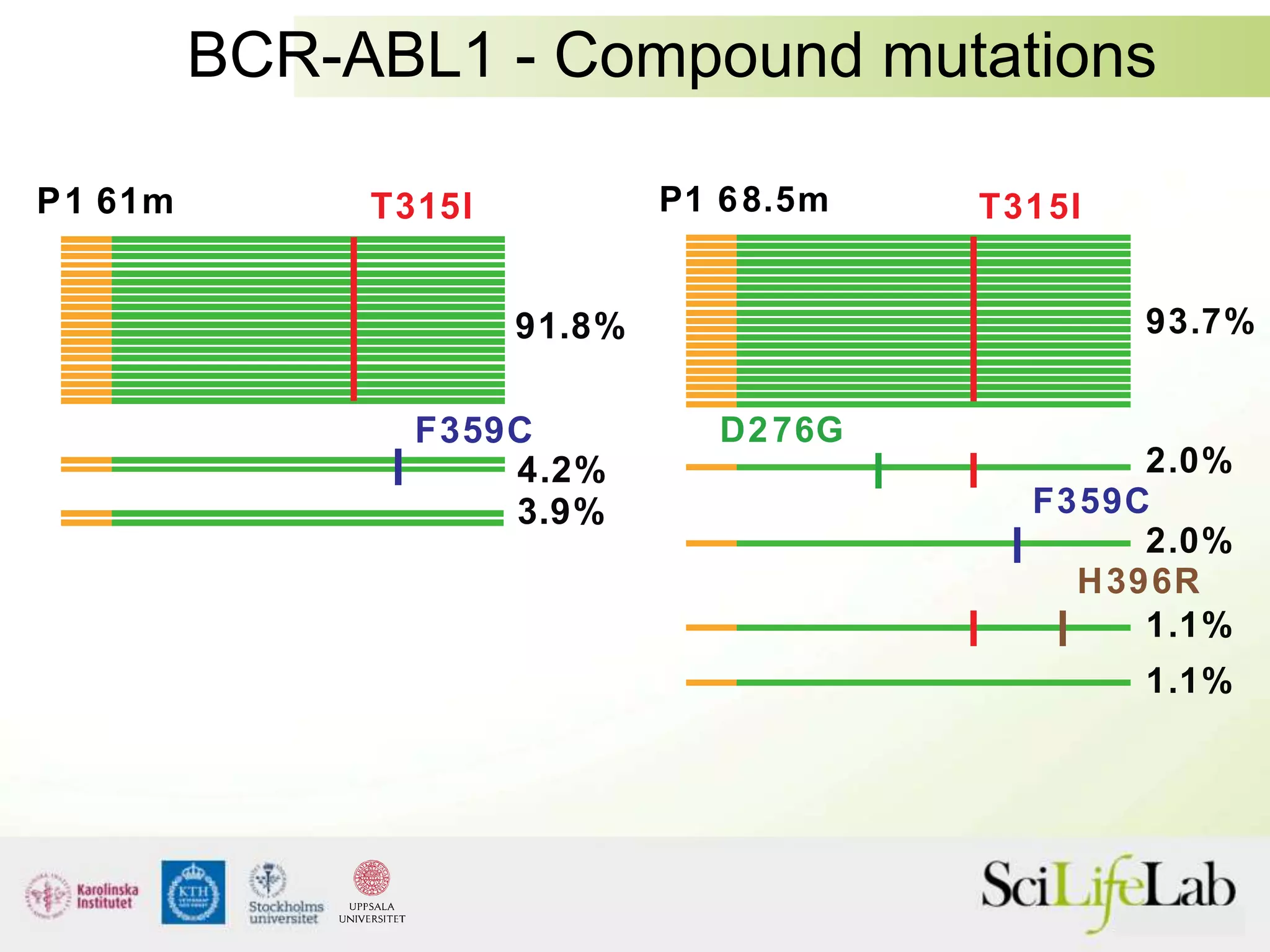
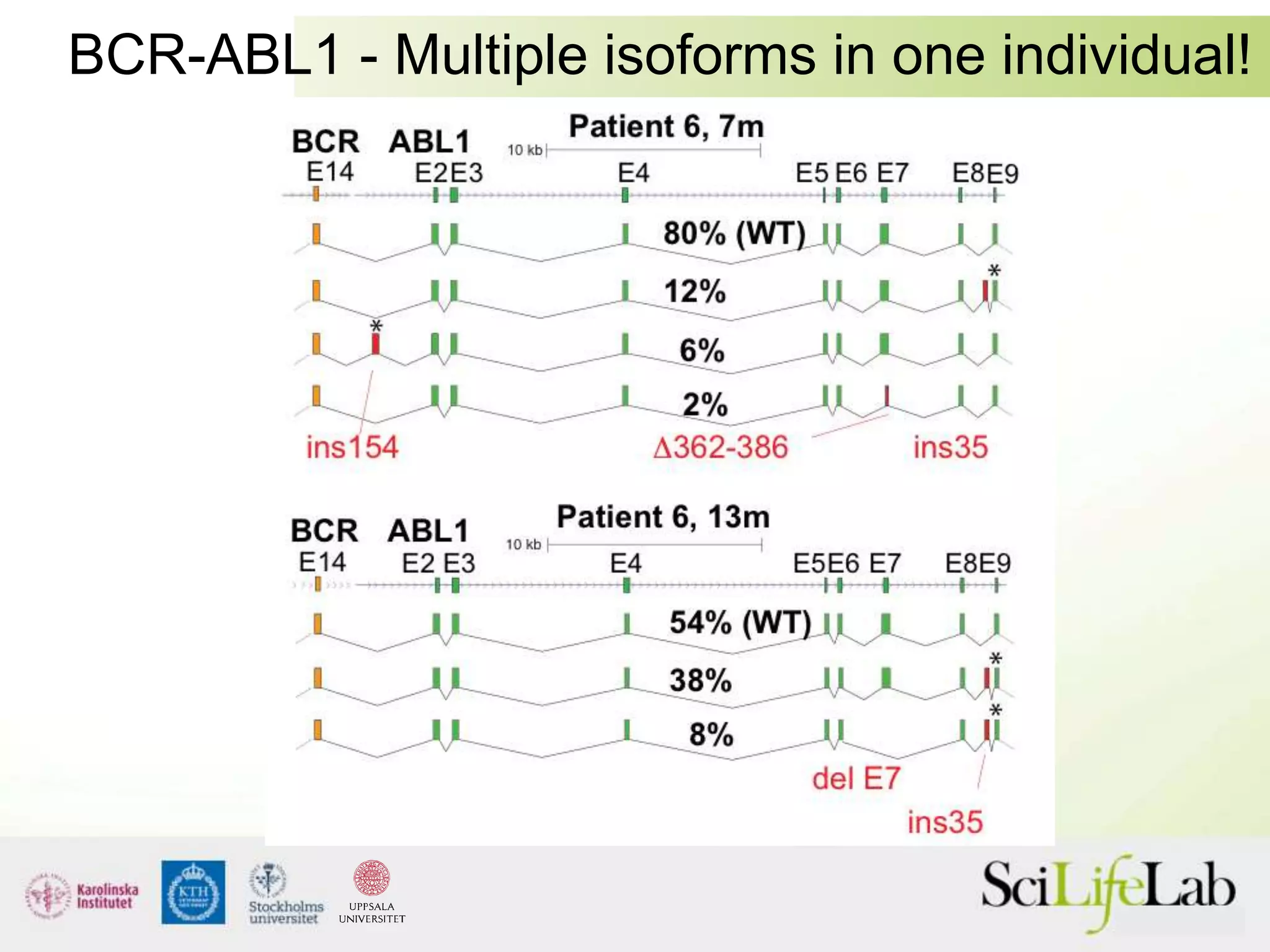
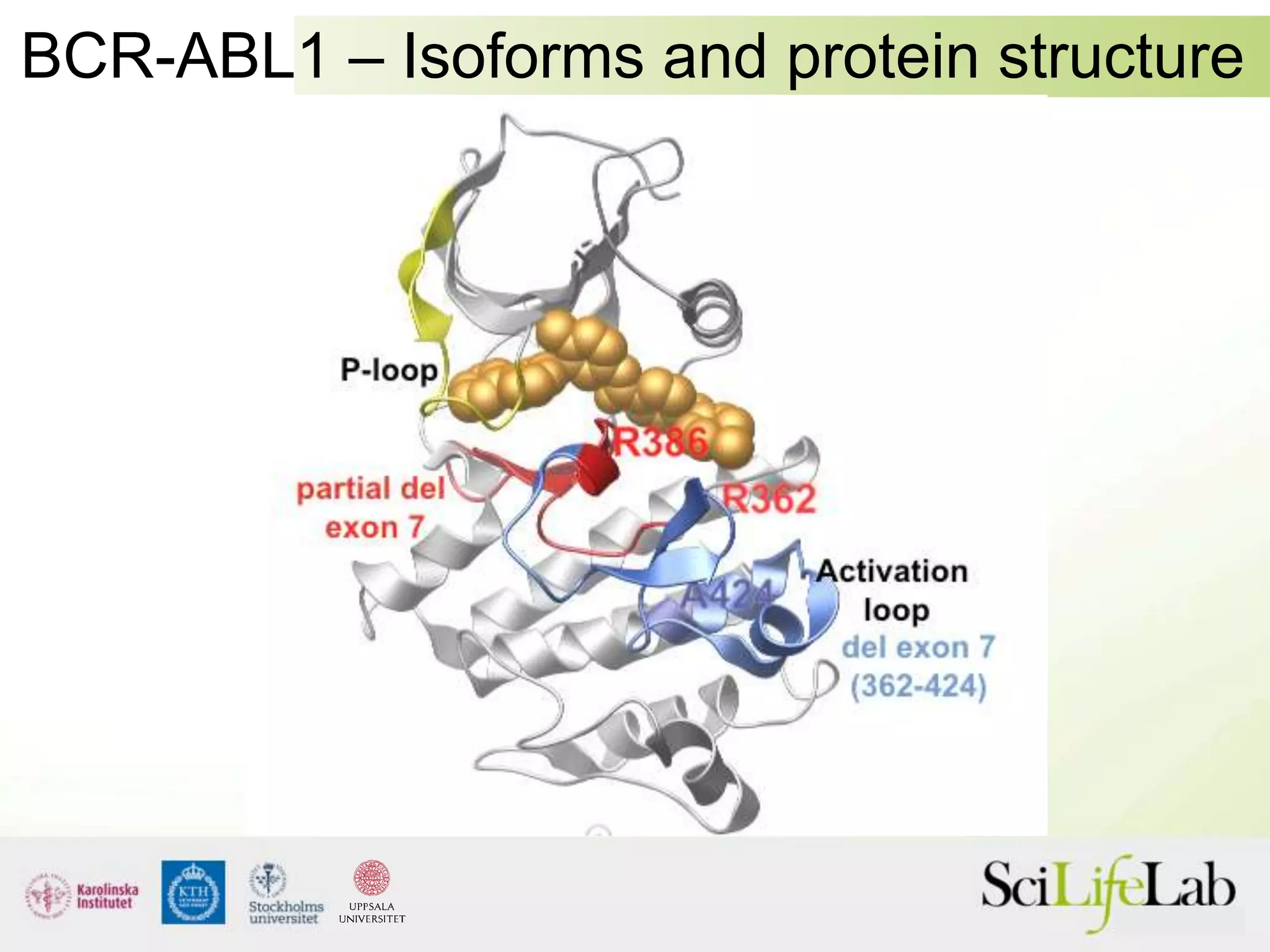
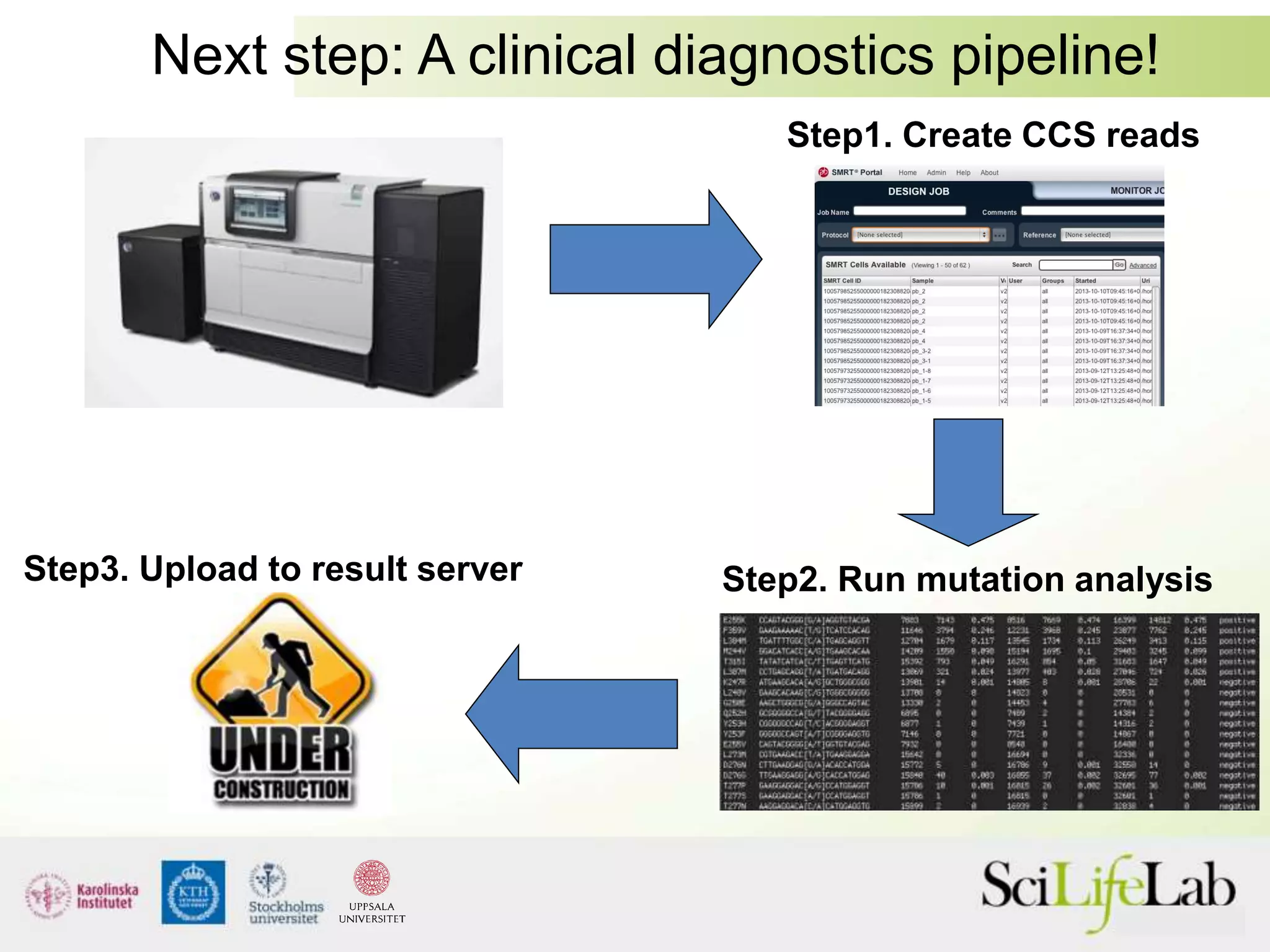
This document summarizes next generation sequencing and bioinformatics analysis pipelines. It discusses the Ion Torrent and Pacific Biosciences sequencing platforms, and provides examples of standard and in-house analysis pipelines for these platforms. It also gives three specific examples of in-house developed pipelines: (1) a local variant database for exome sequencing analysis, (2) de novo genome assembly using long reads from Pacific Biosciences, and (3) a clinical sequencing workflow for detecting mutations in leukemia patients. The document outlines ongoing developments for both sequencing technologies and analysis pipelines.