Downloaded 24 times









![Human Genome Project (15 years) Hierarchical
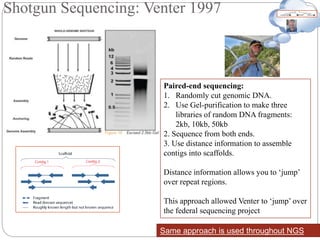
Shotgun Sequencing [start1990]
- Randomly insert Human DNA into BAC clones (~150kbp each)
- Combine these BAC clones to create a scaffold of the human
genome. Each BAC clone will be mapped to a region on a Human
Chromosome
- Pass BAC clones to different Genome Centers throughout US
- At each center, each vector is sequenced using shotgun sequencing
- Wait 15 years for results.](https://image.slidesharecdn.com/hamas1-161205090720/85/Hamas-1-15-320.jpg)


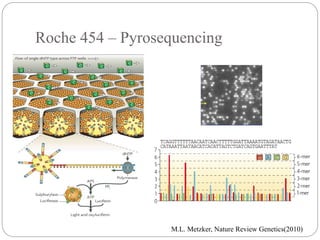
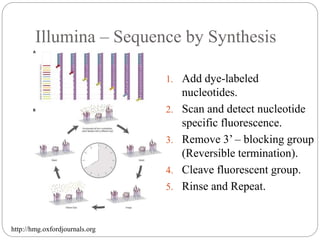
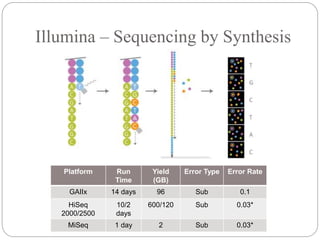


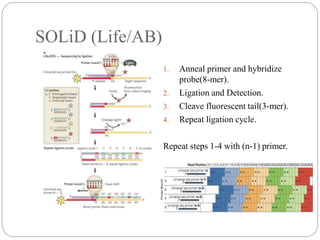

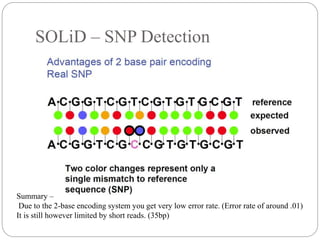
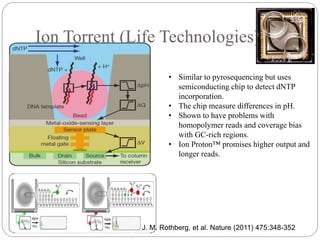
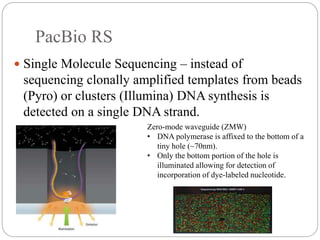

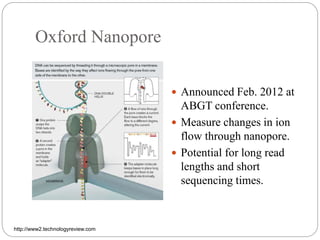

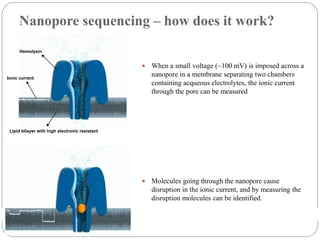
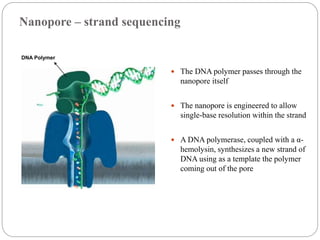

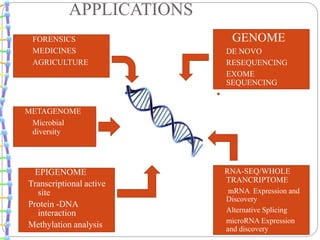



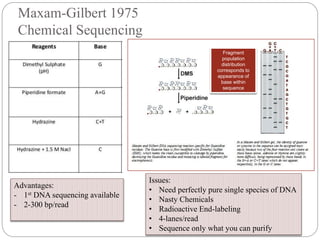
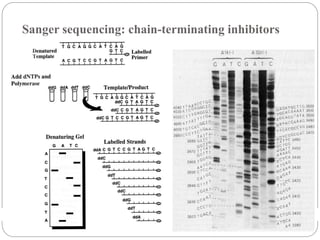
This document summarizes trends in DNA sequencing methods and applications. It discusses the purpose and historical methods of DNA sequencing, including the Maxam-Gilbert and Sanger methods. Next generation sequencing methods like Roche 454, Illumina, SOLiD, Ion Torrent, and PacBio are described. Applications of sequencing include analyzing gene structure, detecting mutations, microbial identification, and whole genome sequencing. The document provides details on sequencing techniques, platforms, yields, and error rates.



























































































![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)







