Downloaded 160 times



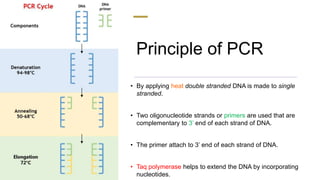
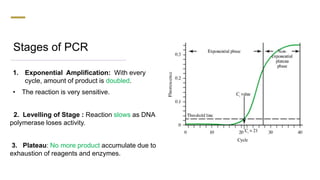

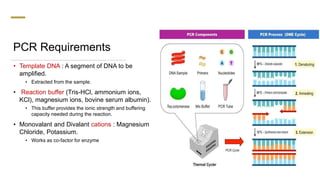







































The document presents an extensive overview of the Polymerase Chain Reaction (PCR), detailing its principles, stages, techniques, modifications, and applications in scientific and clinical settings. It highlights the history of PCR, its requirements, and the various methods used to analyze and amplify DNA sequences effectively. Additionally, it discusses several advanced PCR techniques and their specific uses in research, gene therapy, and diagnostics.
















































































