Downloaded 243 times



















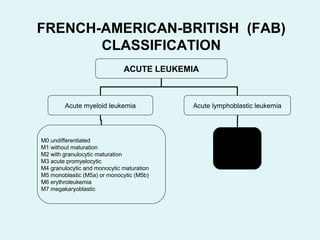



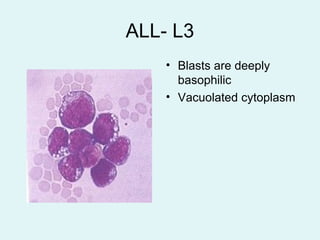


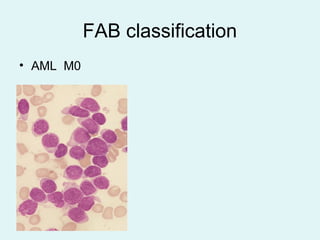


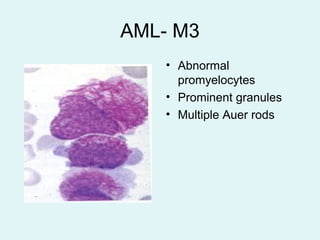
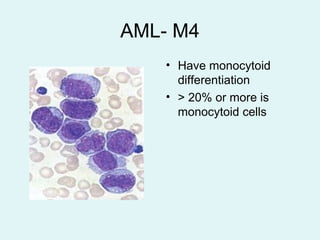


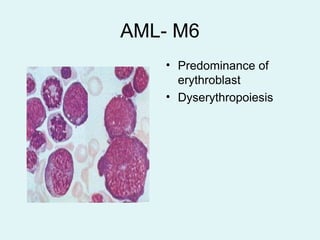
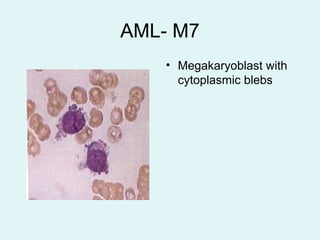






































Leukemias are malignant diseases of the bone marrow characterized by the abnormal proliferation of white blood cells. The document discusses the classification, pathogenesis, epidemiology, clinical features, diagnosis and prognosis of various types of leukemias. It describes the key subtypes of acute and chronic leukemias, focusing on chronic myeloid leukemia, which results from a genetic translocation forming the Philadelphia chromosome and the constitutively active BCR-ABL fusion oncogene that drives uncontrolled myeloid cell growth.























































































