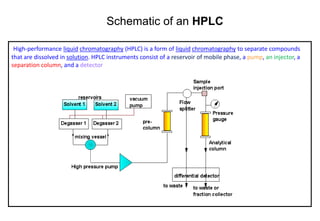
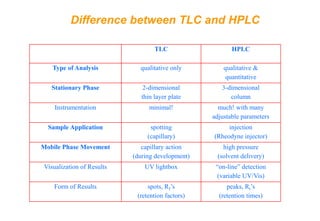
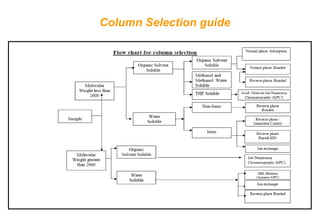
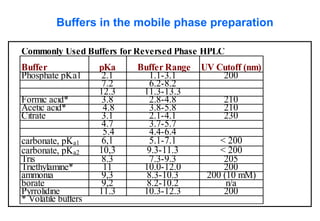
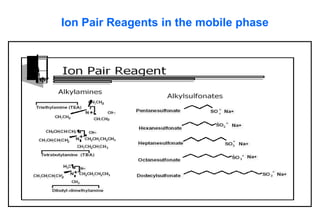
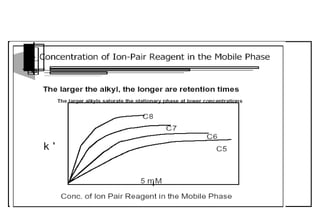
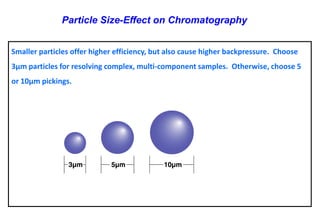
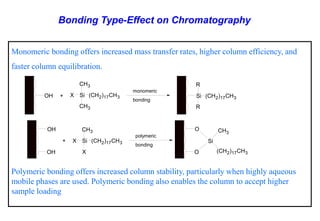
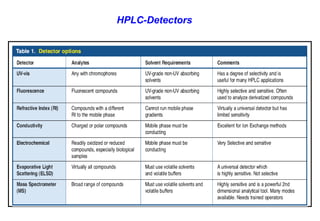
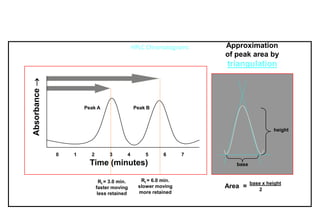
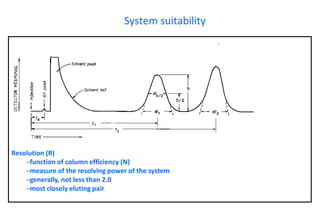
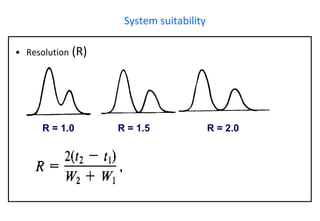
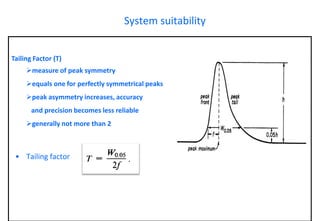
This document provides information about handling and operating high performance liquid chromatography (HPLC). It discusses the basic components and setup of an HPLC instrument. It also compares HPLC to thin layer chromatography. The document explains reverse phase and normal phase HPLC, proposed mechanisms, column selection, mobile phase preparation including buffers, ion pair reagents, column properties, and common detectors. It provides guidelines for handling HPLC including preparation of samples, standards and mobile phases, and assessing system suitability.