• Hemo =means blood
• stasis = arrest or stoppage
• Hemostasis = is the process of
Stoppage of bleeding from a
damaged blood vessel.
Hemostasis
4.
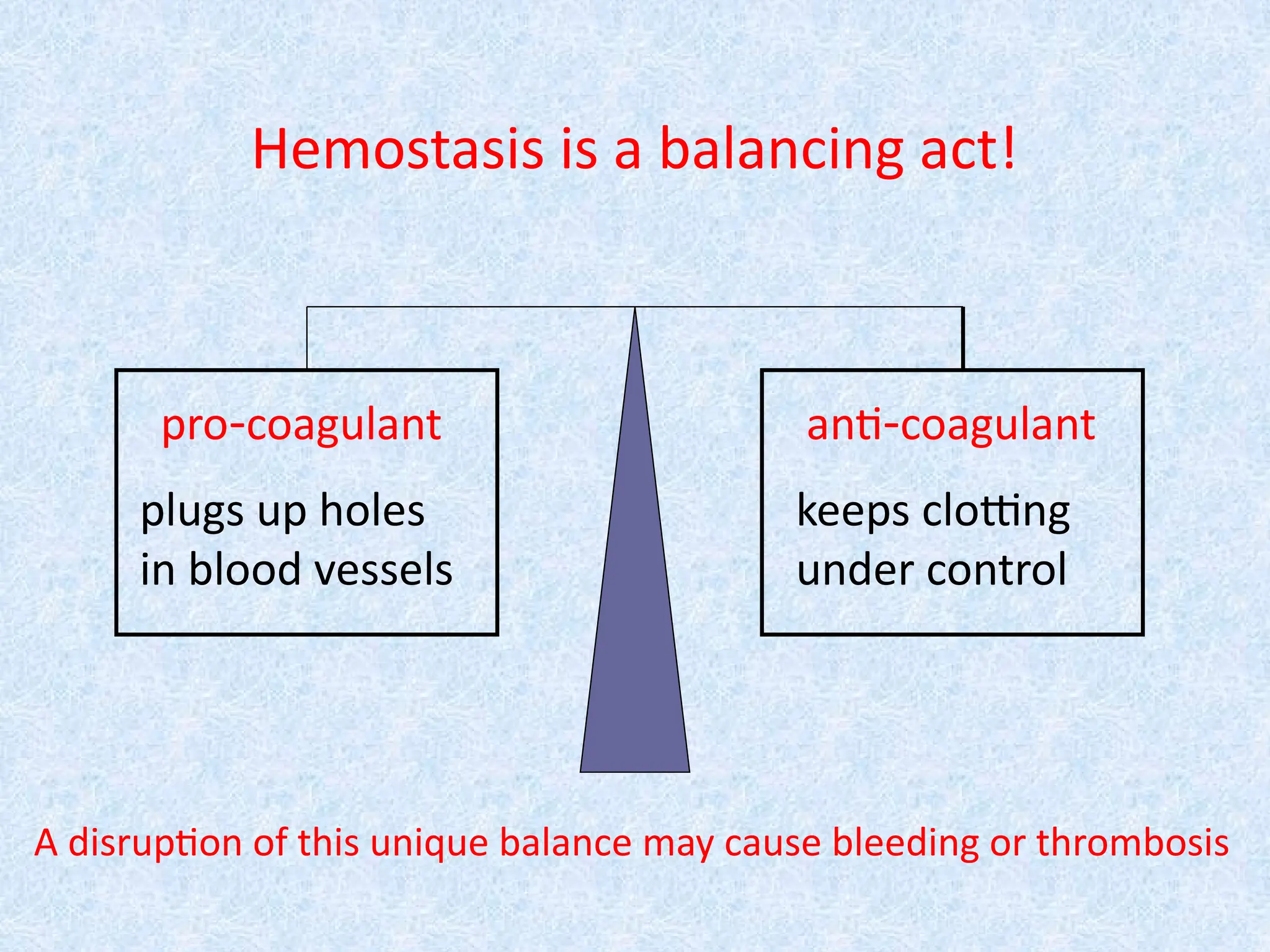
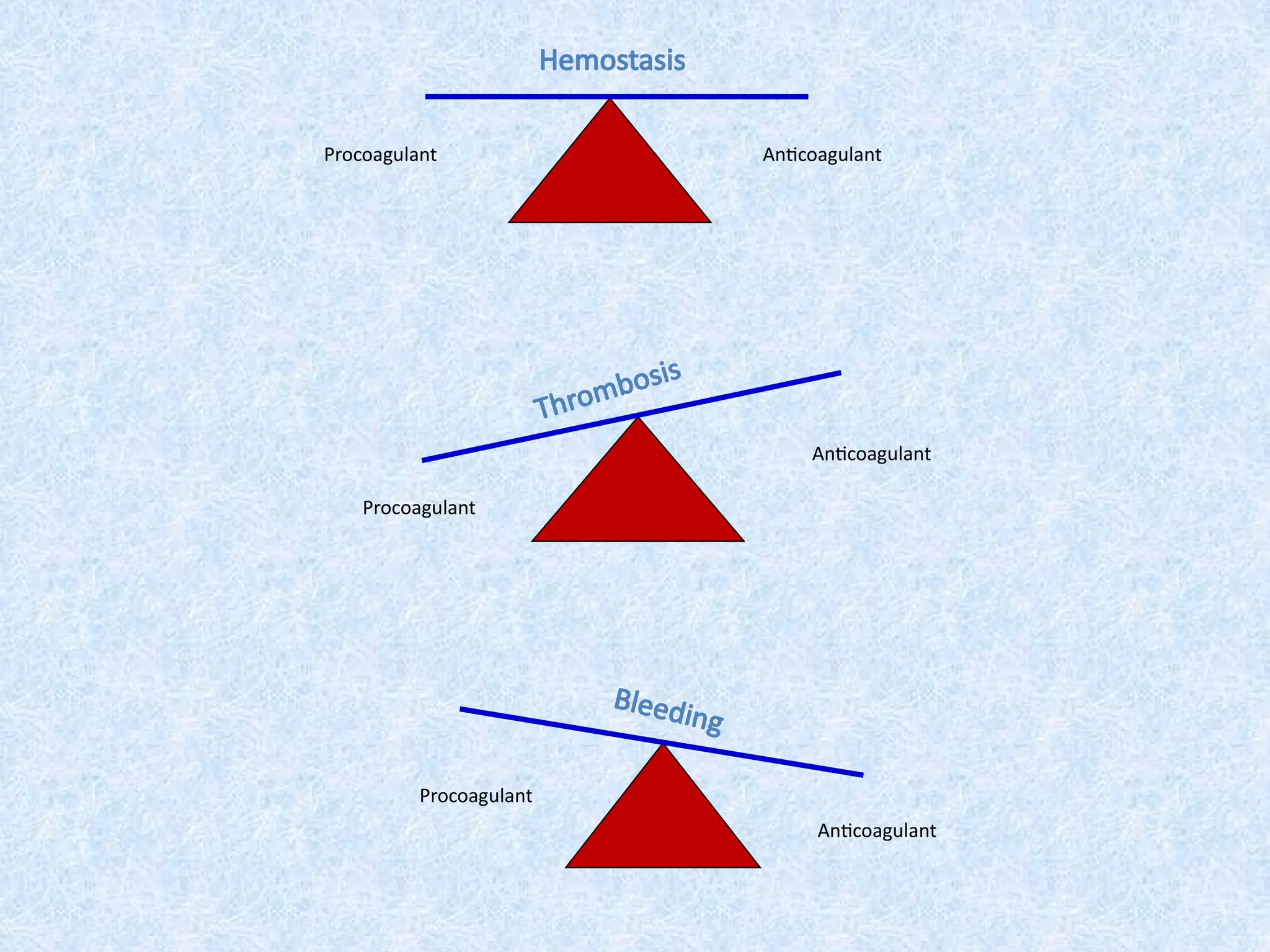
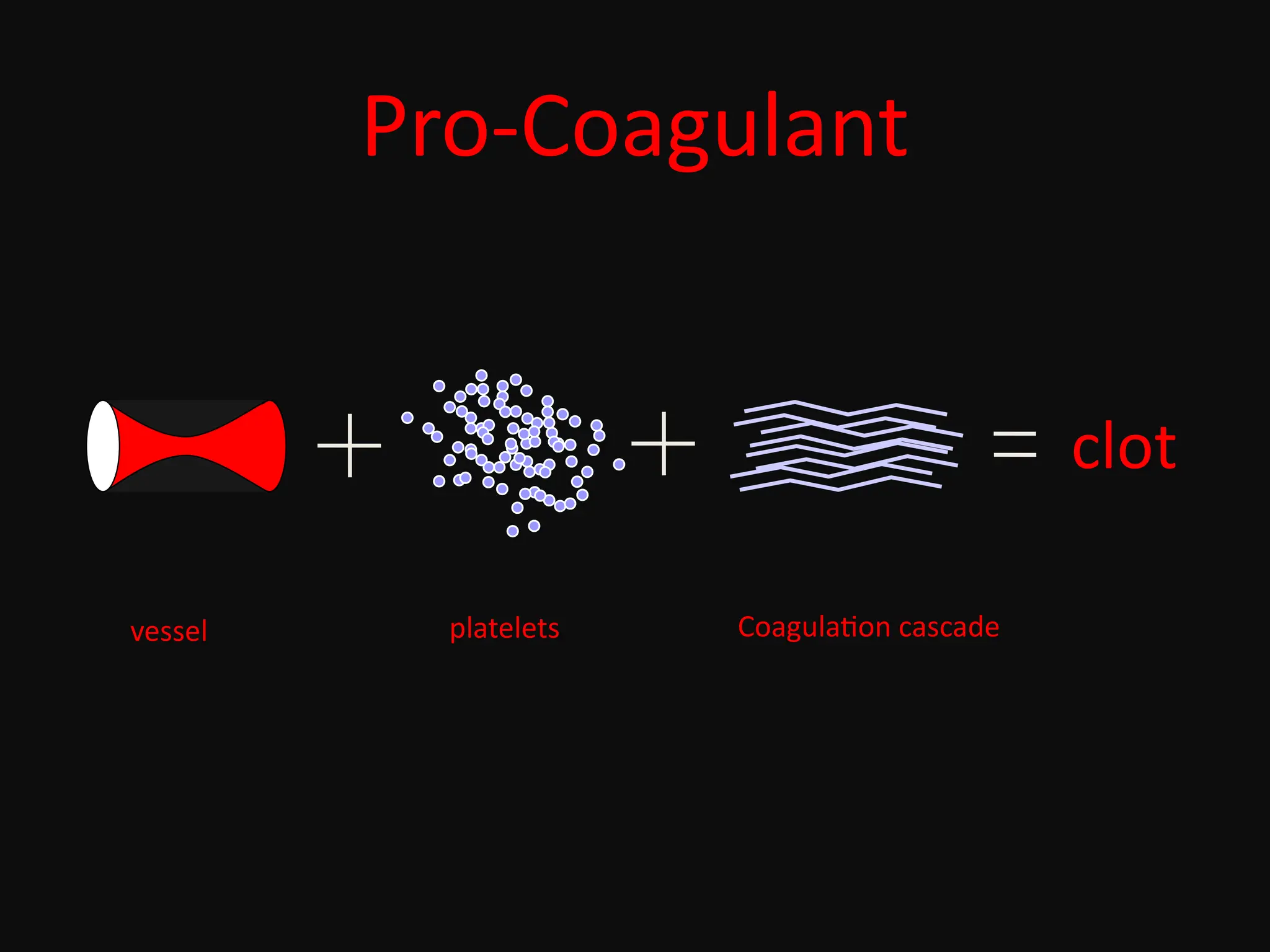
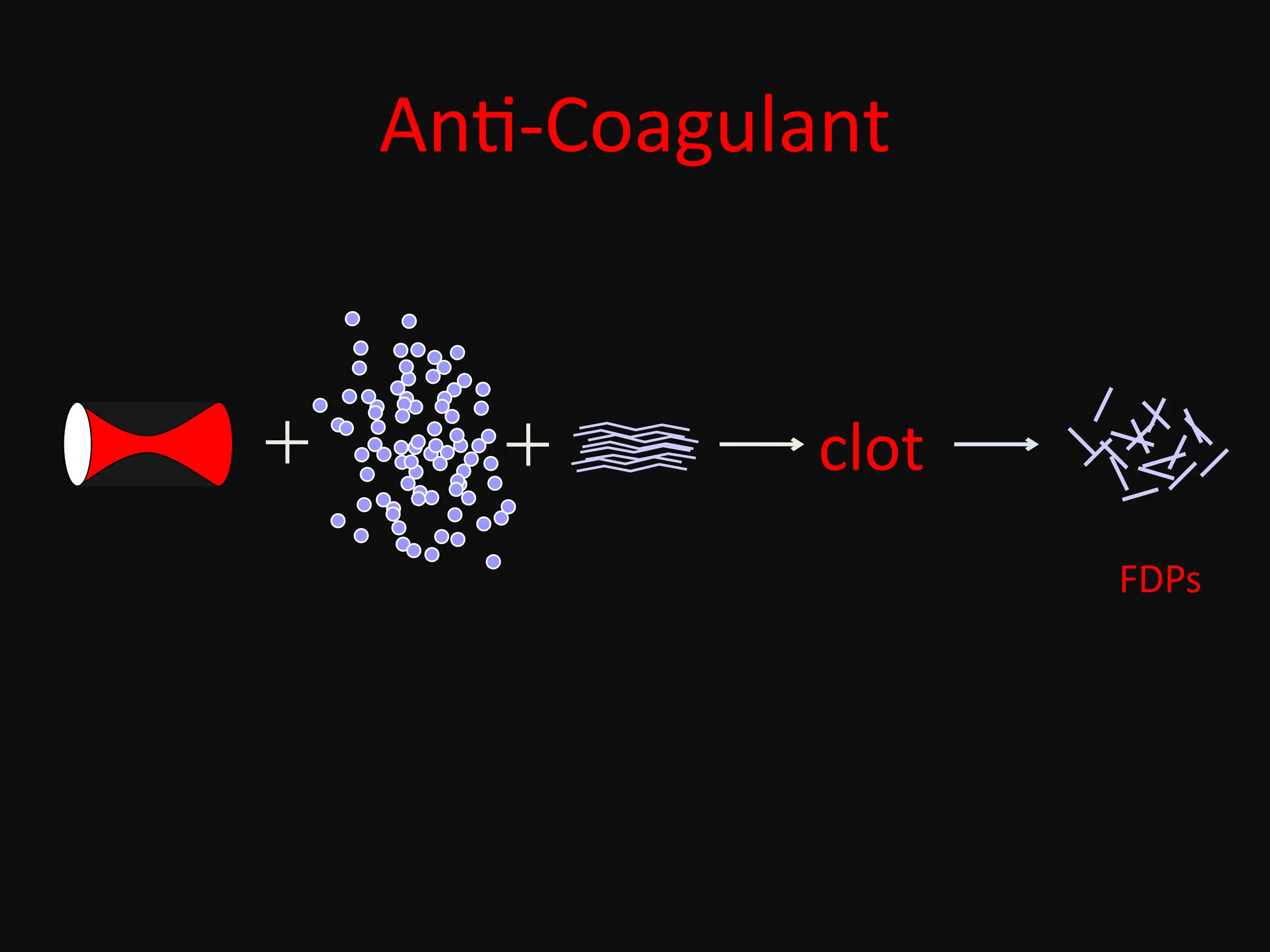
Hemostasis is abalancing act!
pro-coagulant anti-coagulant
plugs up holes
in blood vessels
keeps clotting
under control
A disruption of this unique balance may cause bleeding or thrombosis
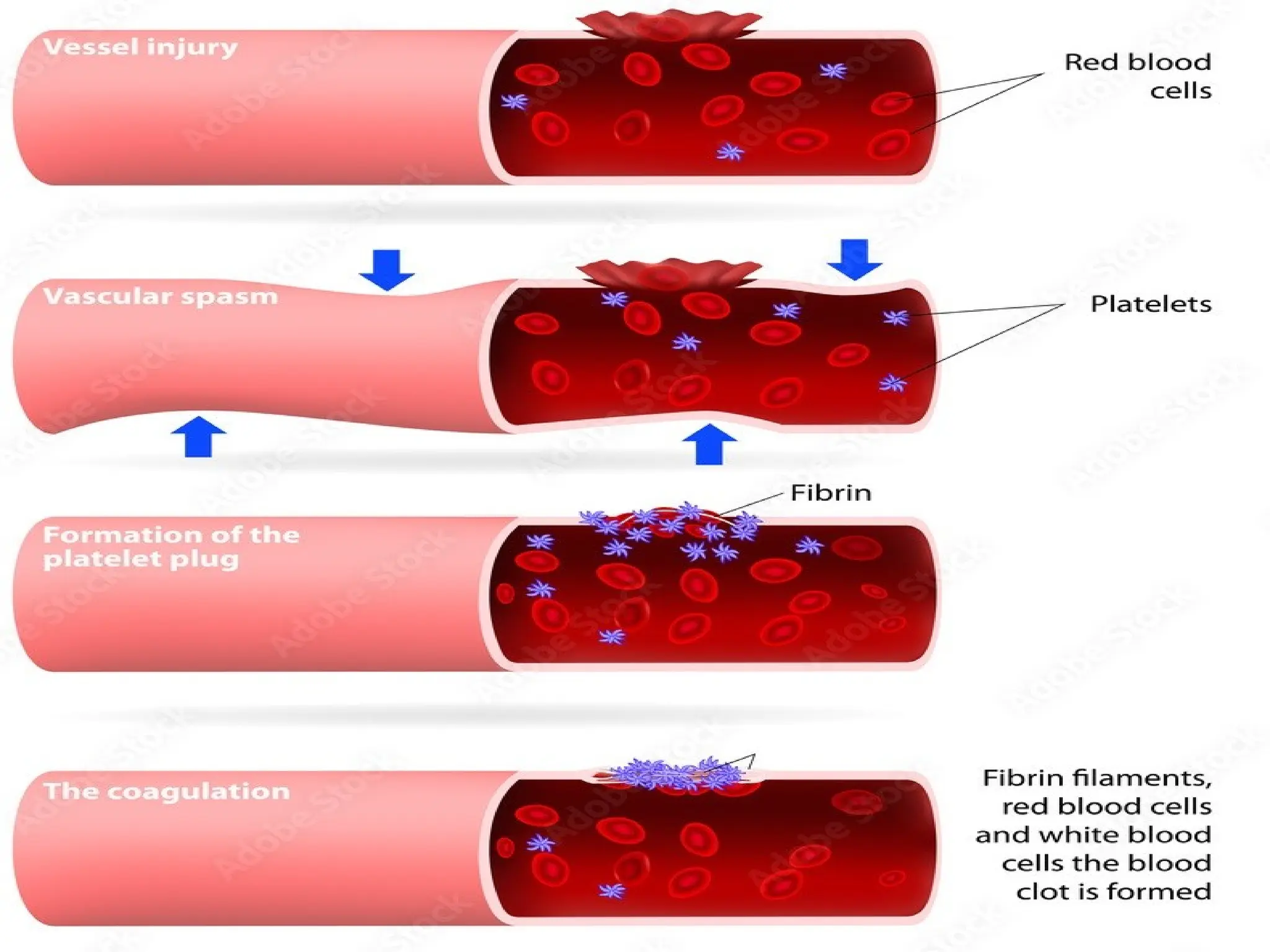
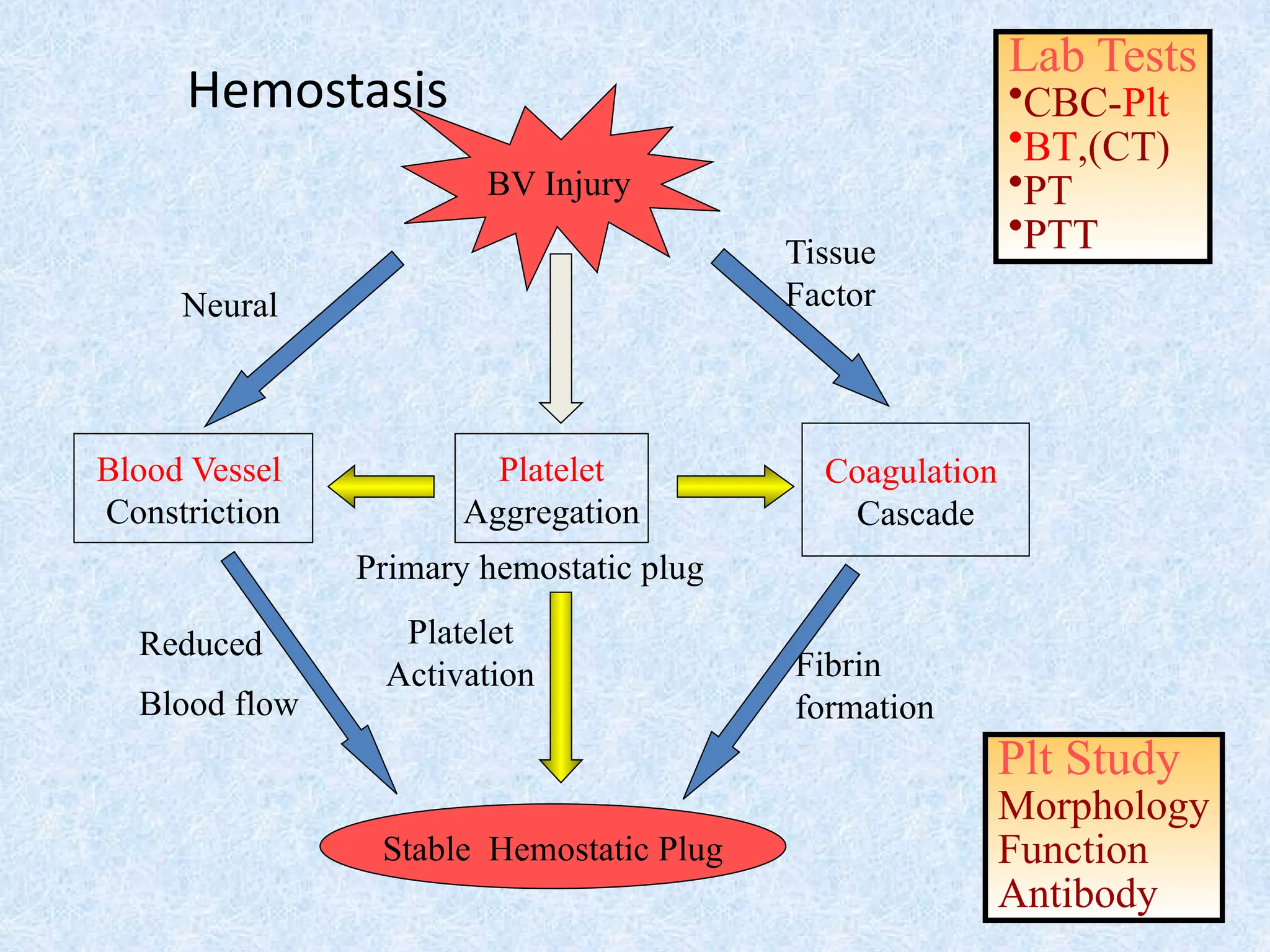
• Fast andlocalized reaction when a
blood vessel breaks.
• Involves a series of reactions.
• Involves substances normally found in
plasma but not activated.
• Occurs in 4 main phases
Hemostasis
Coagulation disorders (Coagulopathy)
Acquiredcoagulation disorders
1. Disseminated intravascular coagulation
2. Liver disease
3. Vitamin K deficiency
4. Acquired inhibitors of coagulation
5. Heparin, oral anticoagulation, thrombolytic therapy
6. Renal disease
7. Paraproteinaemias
8. Cardiopulmonary bypass
9. Massive transfusion of stored blood
Inherited coagulation disorders
1. hemophilia A (Factor VIII deficiency)
2. hemophilia B (Factor IX deficiency)
3. von Willebrand disease.
26.
Evaluation of ableeding patient
• Spontaneous bleeding or related to trauma?
o Spontaneous = platelet or vascular defect
o Related to trauma = coagulation defect
• Site of bleeding ? (superficial or deep)
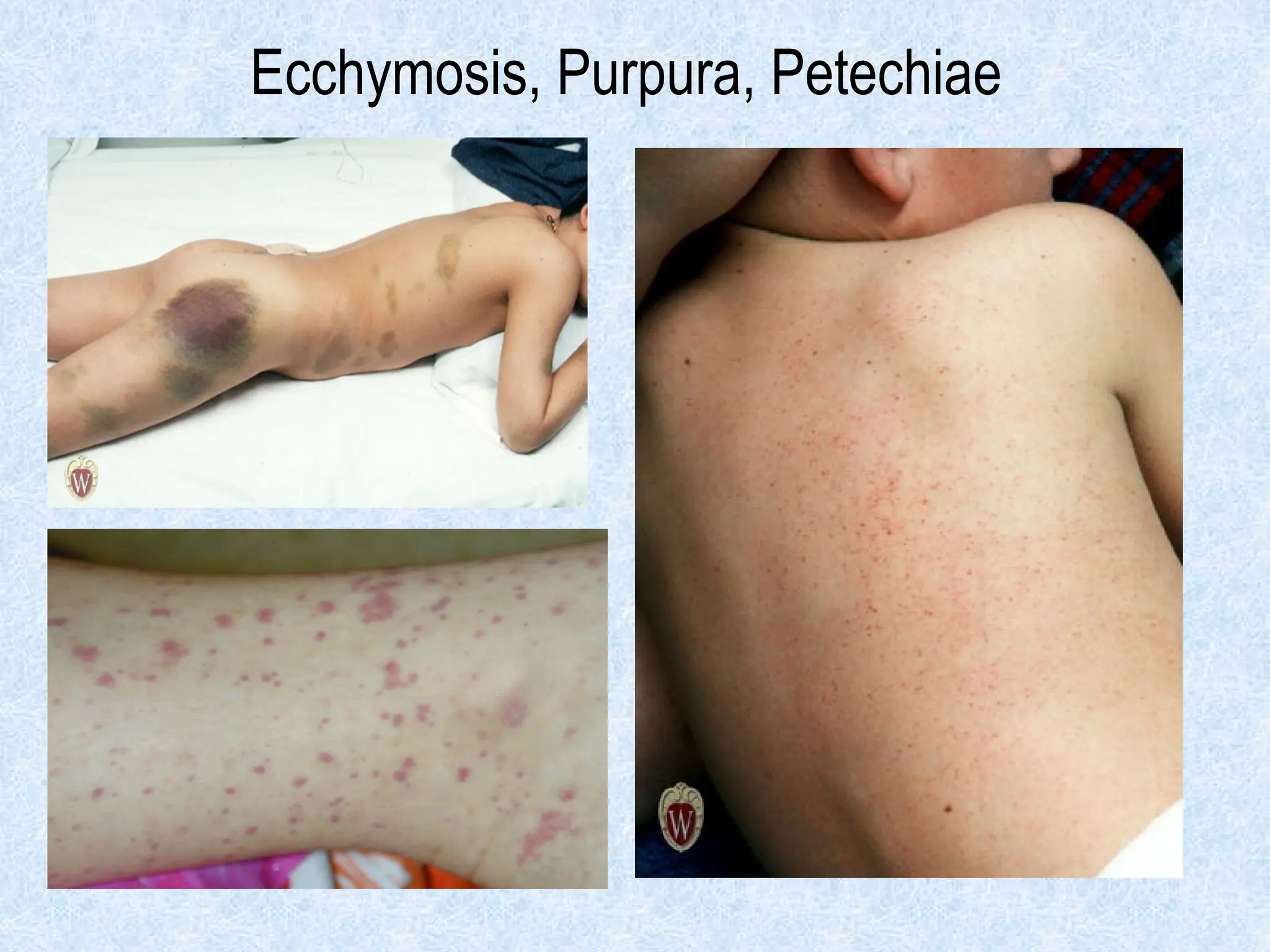
o Superficial bleeding:
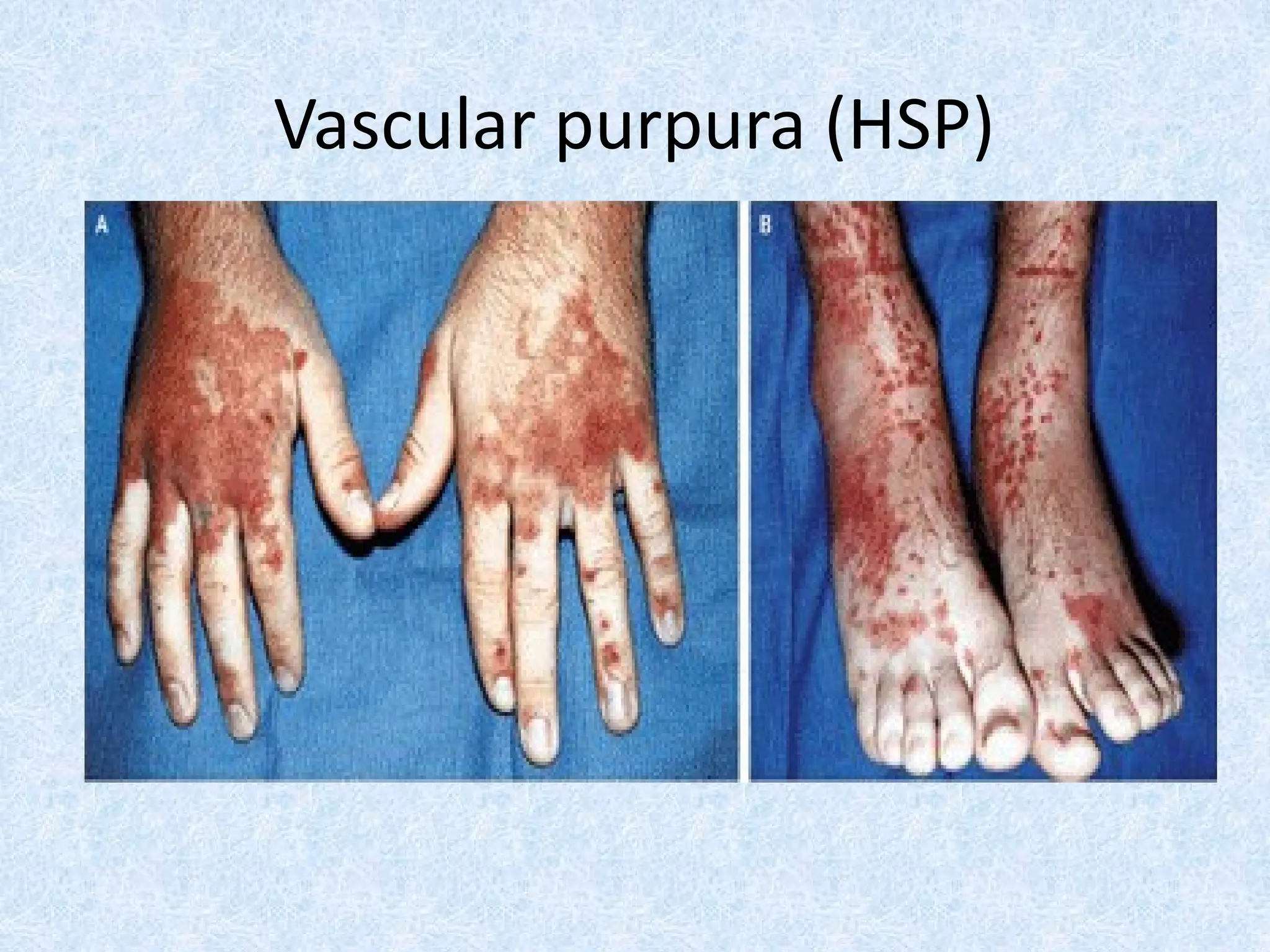
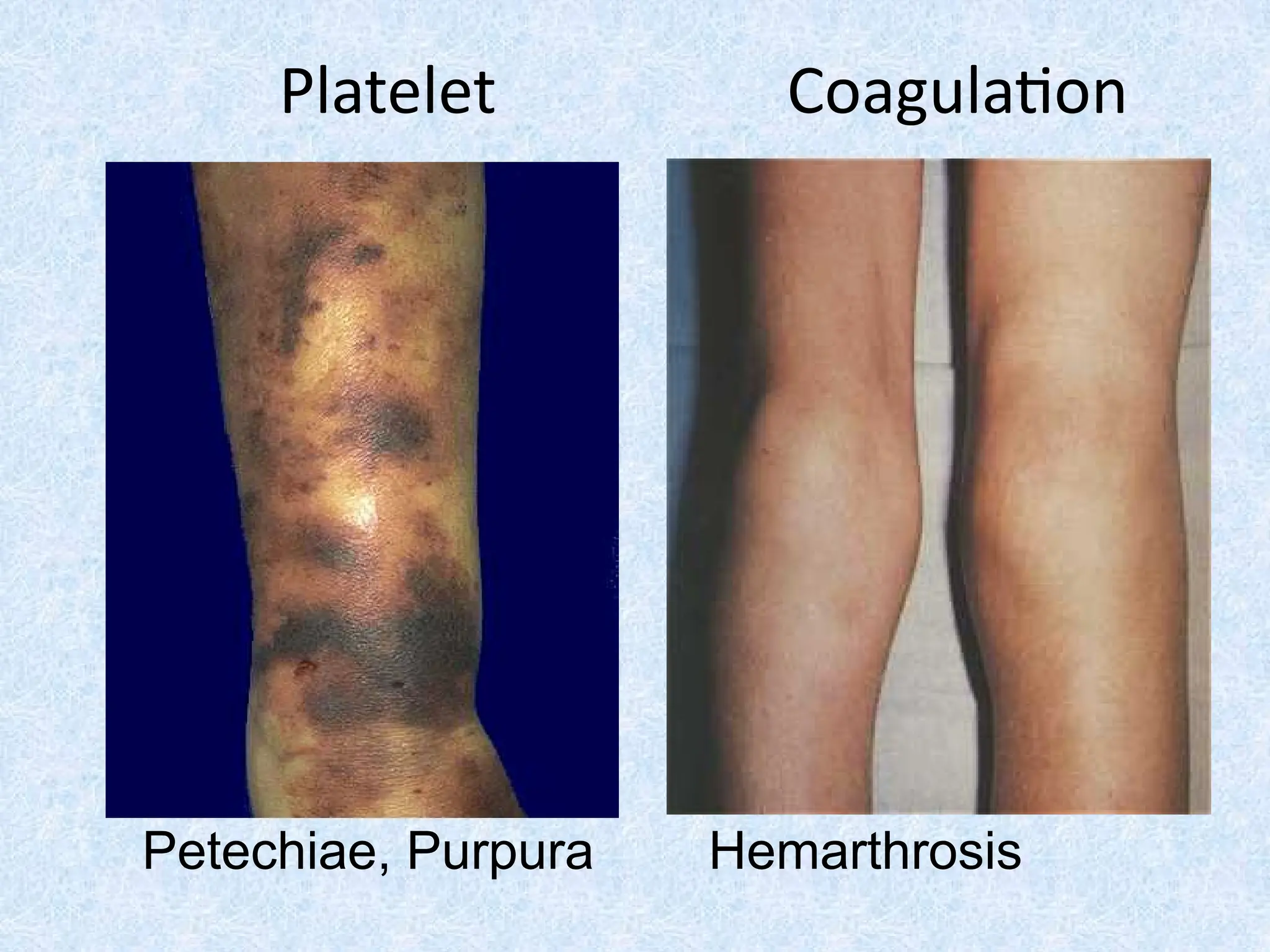
Skin bleeding (petechiae, purpura, ecchymosis)
Menorrhagia
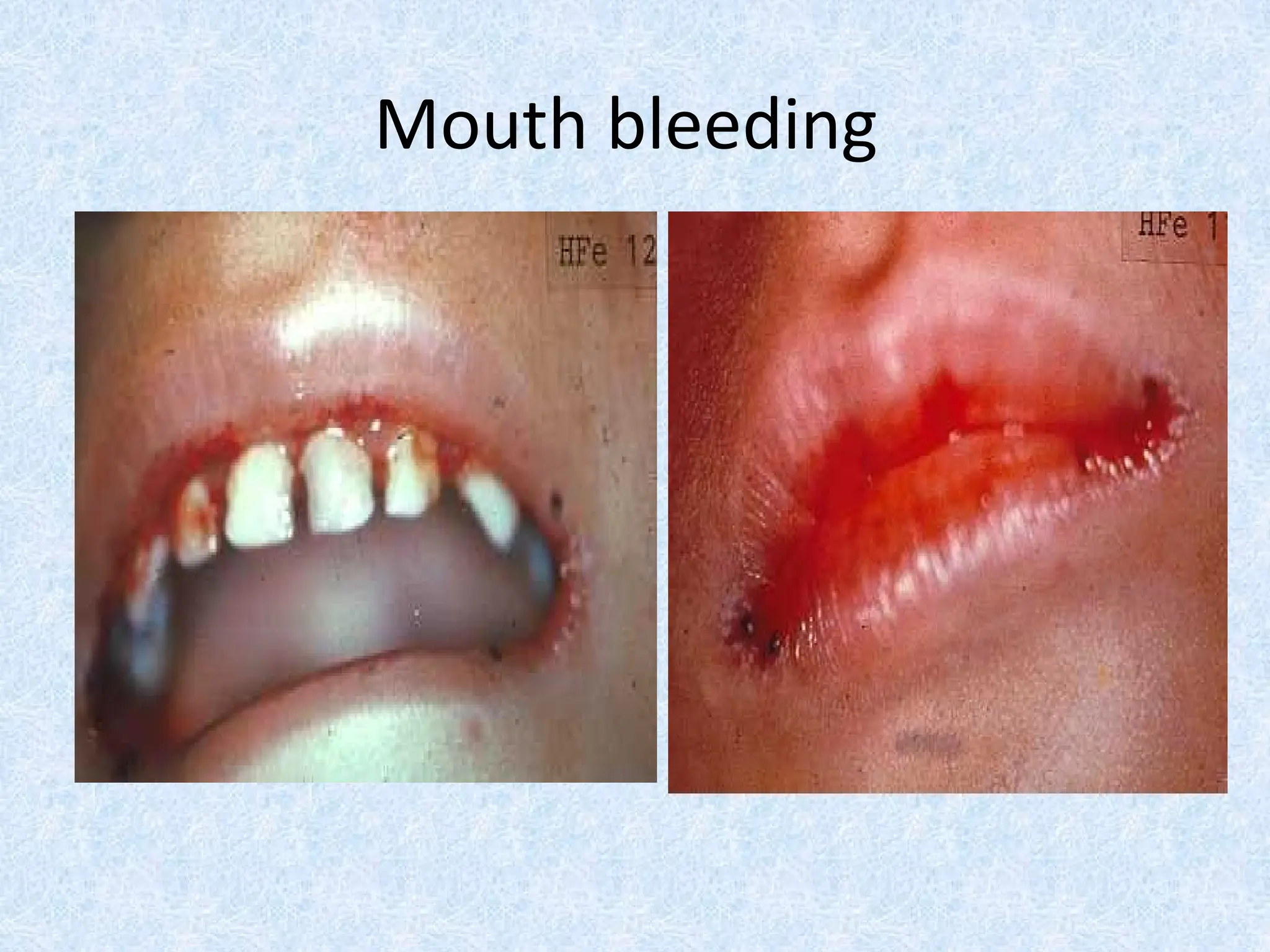
Mouth bleeding (gum bleeding)
Epistaxis
Bleeding per rectum
Hematuria
o Deep bleeding:
Joint bleeding (Hemarthrosis)
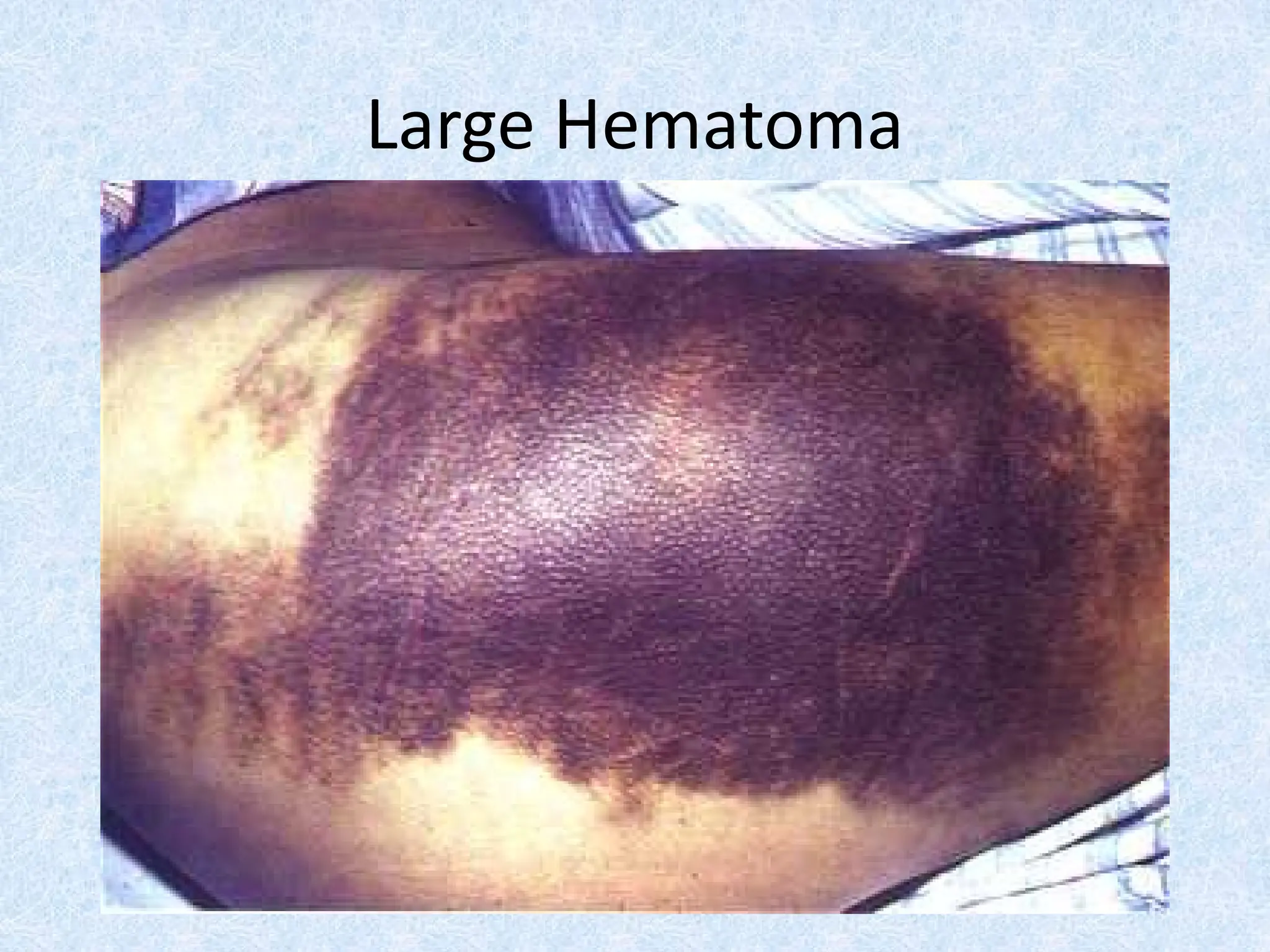
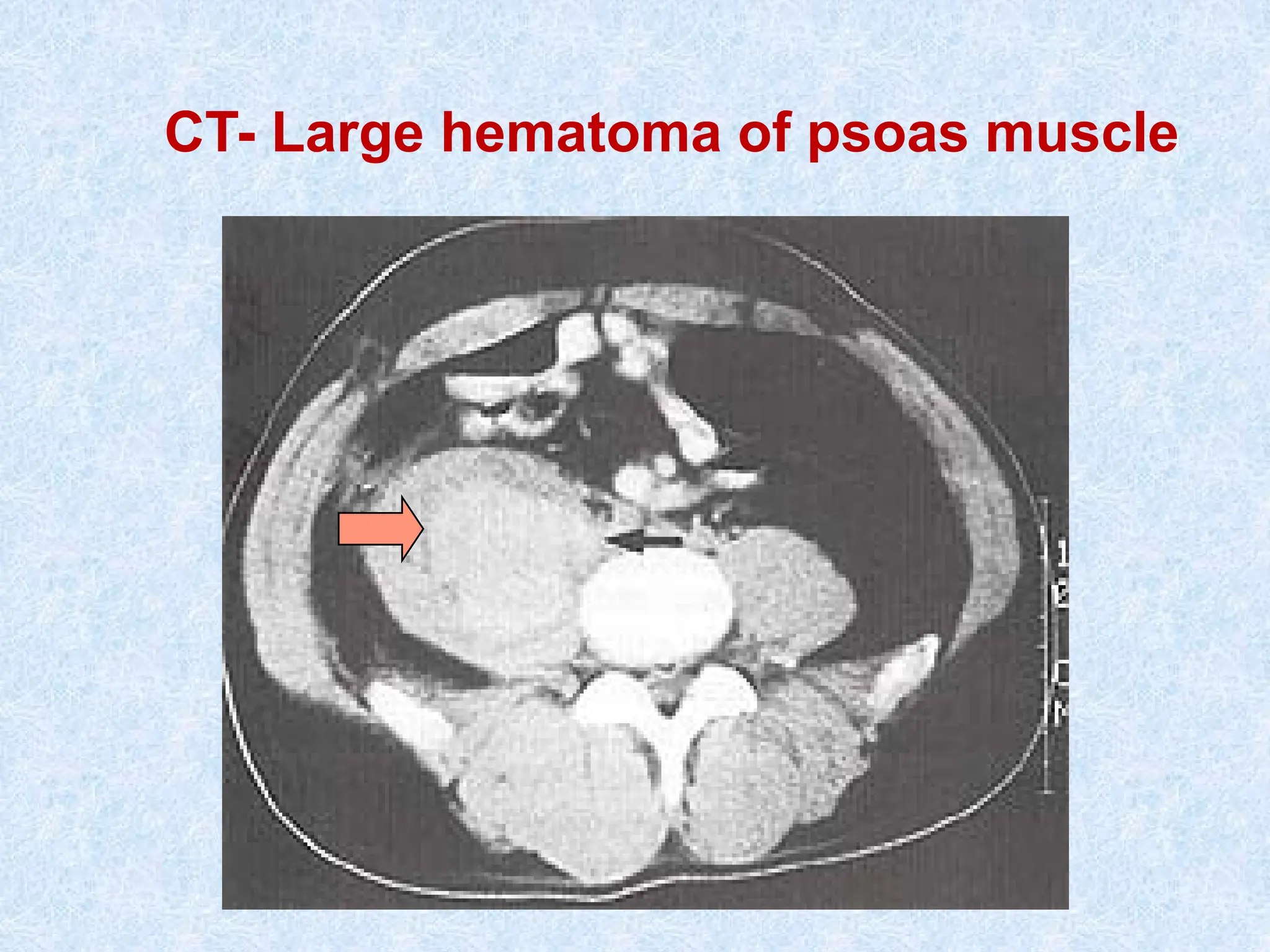
Muscle hematoma (psoas ms hematoma)
Intracavitary he (Intracranial hge)
o Superficial bleeding = platelet or vascular defect
o Deep bleeding = coagulation defect
27.
• Time aftertrauma ?
o Immediate bleeding = Platelet or vascular defect
o Delayed bleeding = Coagulation defect
• Onset of bleeding ?
o At childbirth, early childhood = congenital ds
o At adulthood = acquired ds
• Existent comorbidities e.g., liver, renal, malignancy,..
• Drug history e.g., NSAIDs, Antiplatelets, Antibiotics,..
• Nutritional status (Malnutrition): decreased hepatic synthesis
• Family history ?
o Positive FH = Inherited ds
o Negative FH = Acquired
• Significant bleeding or not ?
o Repeated episodes
o Multiple sites of bleeding
o Requiring transfusion support
o Positive family Hx
28.
Platelet factor Coagulationdisorders
disorders
Site of bleeding Skin Deep in soft tissues
• Mucous membranes (joints, muscles)
• (epistaxis, gum, vaginal,
• GI tract)
Petechiae, purpura Yes No
• Ecchymoses (“bruises”) Small, superficial Large, deep
• Hemarthrosis / muscle bleeding Extremely rare Common
• Bleeding after cuts & scratches Yes No
• Bleeding after surgery or trauma Immediate,
Delayed (1-2 days),
• usually mild often severe
• Prolonged bleeding time prolonged
coagulation time
Laboratory evaluation forpatient with
bleeding
Screening tests: done for all suspected patients
⚫Platelet count
⚫Bleeding time
⚫Prothrombin time (PT)
⚫Partial thromboplastin time (PTT)
⚫Thrombin time (TT)
Specific tests : done according to results of
screening tests
37.
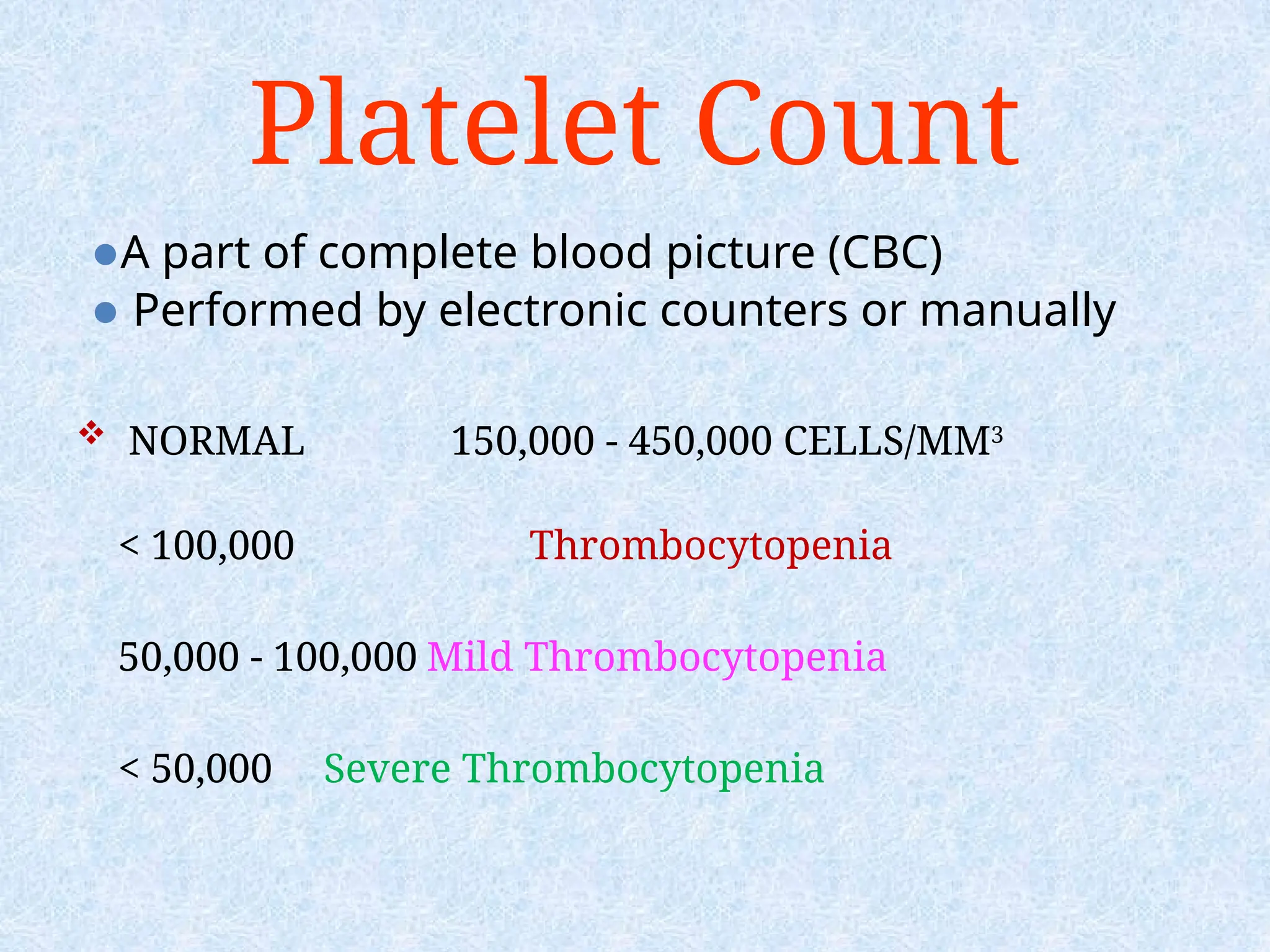
Platelet Count
⚫A partof complete blood picture (CBC)
⚫ Performed by electronic counters or manually
NORMAL 150,000 - 450,000 CELLS/MM3
< 100,000 Thrombocytopenia
50,000 - 100,000 Mild Thrombocytopenia
< 50,000 Severe Thrombocytopenia
38.
Bleeding Time
Time neededto stop bleeding after induced needle
prick
Affected by:
•Platelet count
•Platelet function
•Vessel wall
Normal range: : 2-8 min
Note: it is not a sensitive test for minor or even
moderate abnormality.
39.
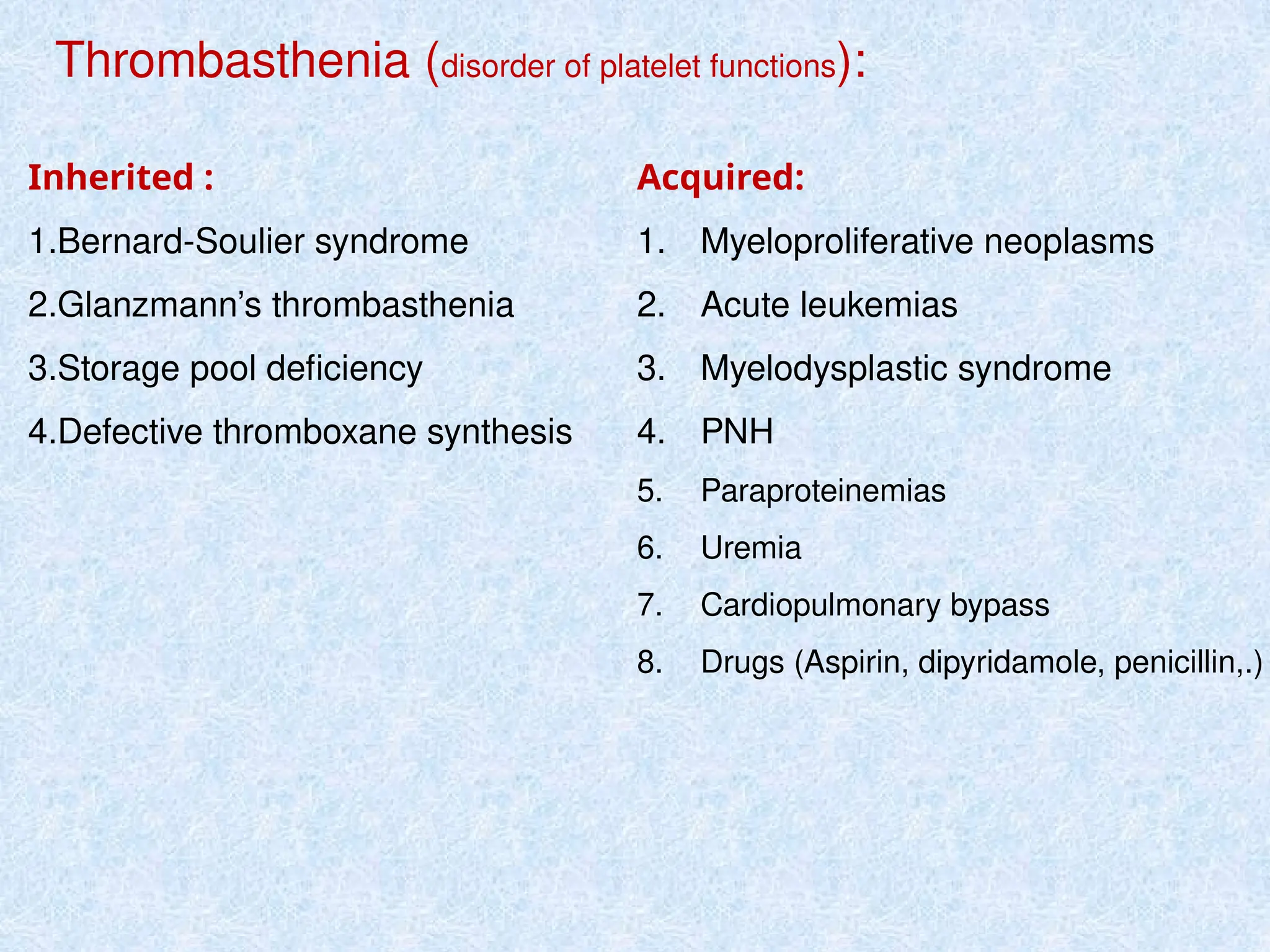
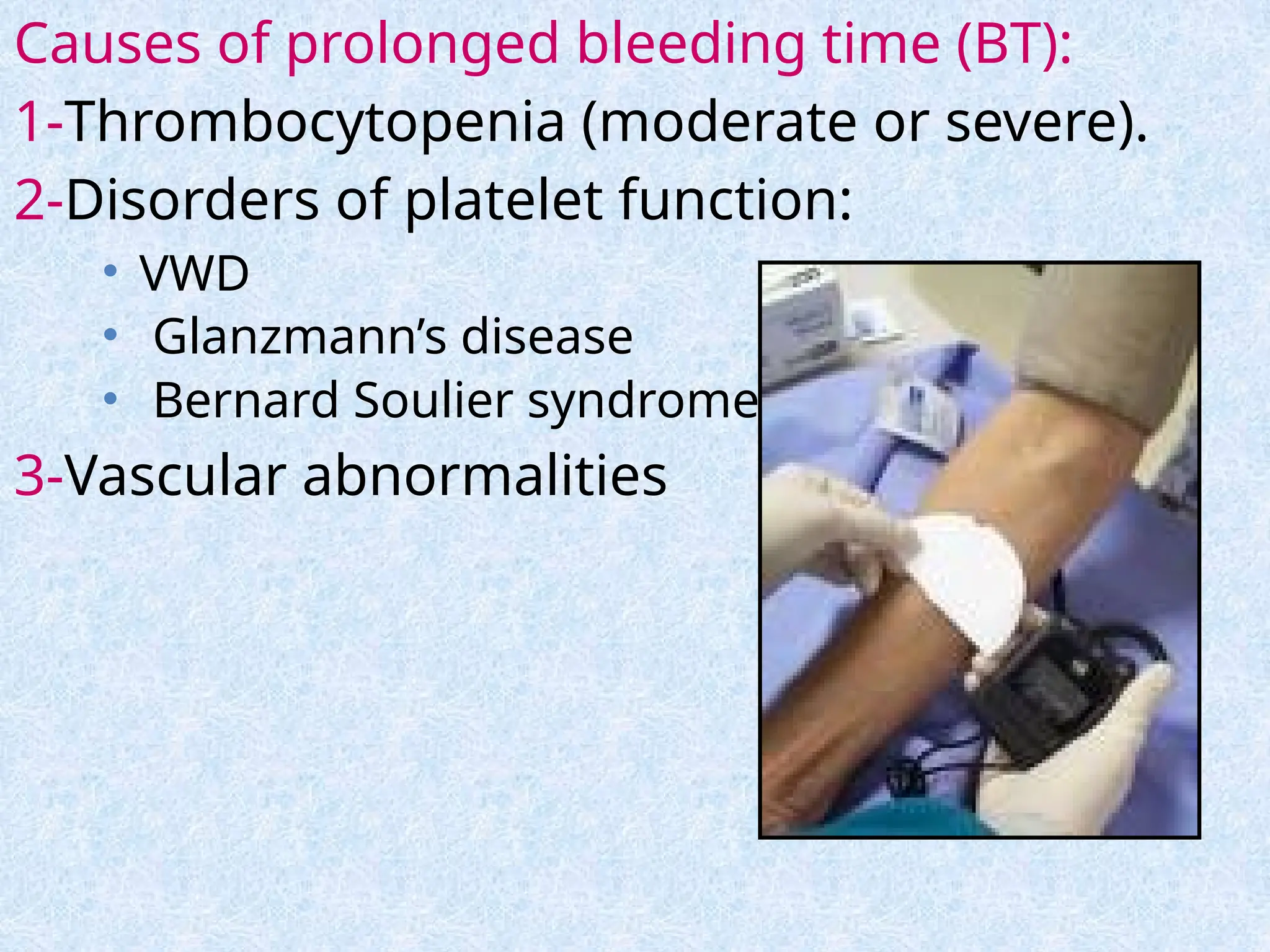
Causes of prolongedbleeding time (BT):
1-Thrombocytopenia (moderate or severe).
2-Disorders of platelet function:
• VWD
• Glanzmann’s disease
• Bernard Soulier syndrome
3-Vascular abnormalities
40.
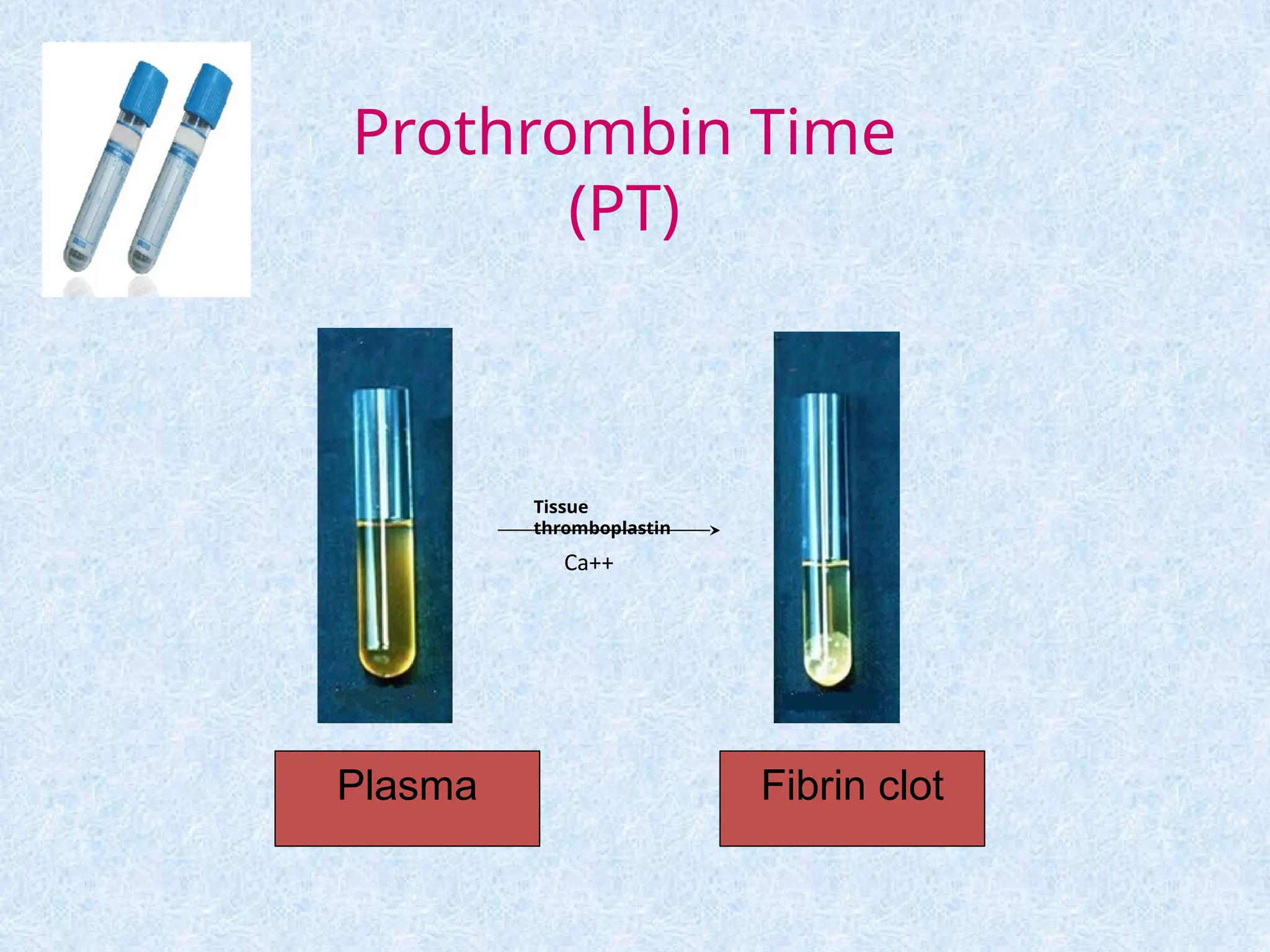
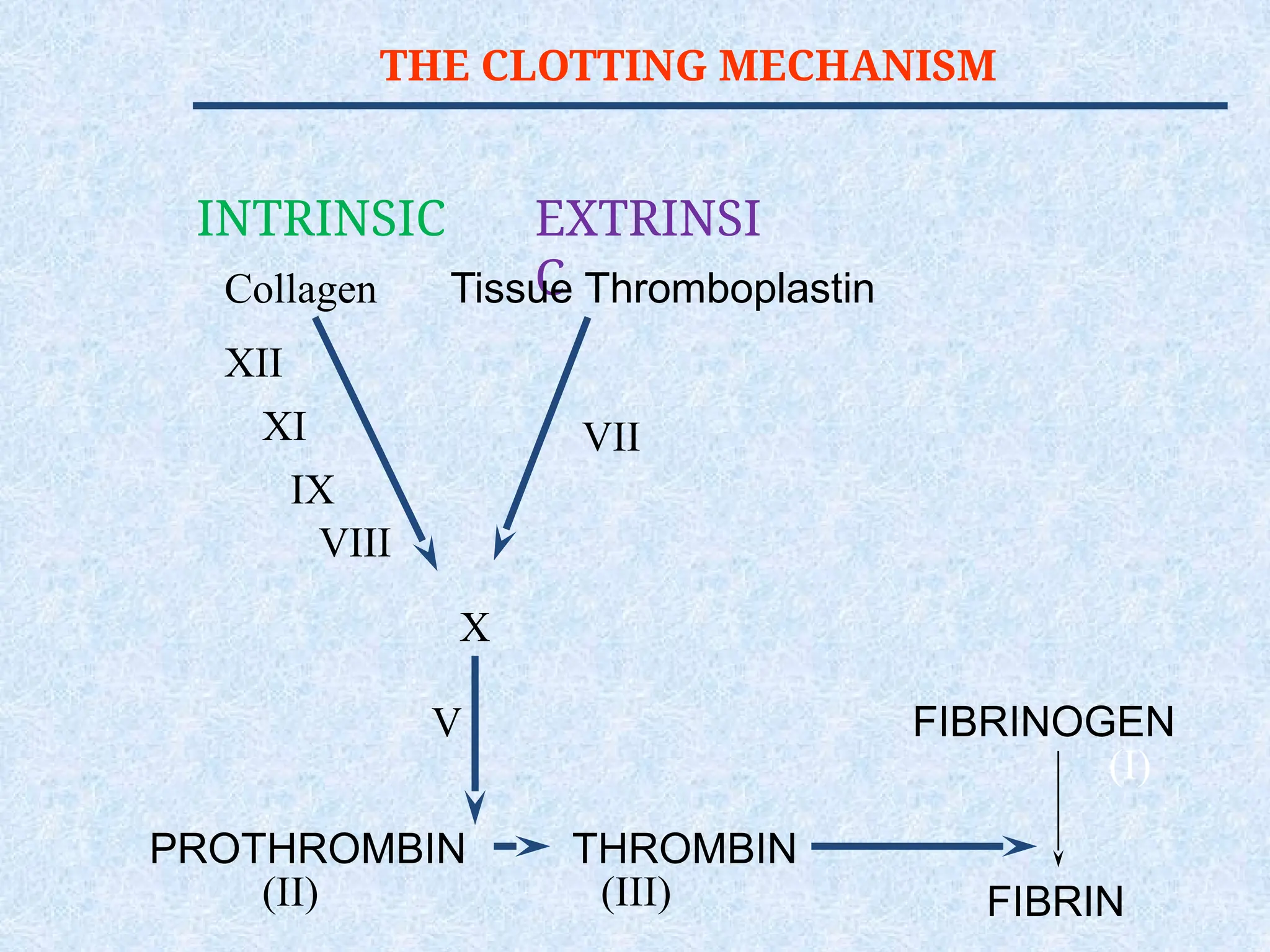
Prothrombin Time (PT)
Measures The Effectiveness of the
Extrinsic Pathway (FVII, FII, FV, X)
[majority are vitamin K dependent
factor].
Used for monitoring oral anticoagulant
therapy and used as a liver function
test
Normal range: 10-14 sec
Causes of
prolonged PT
1.Oral anticoagulants (Warfarin)
2. Liver disease
3. Vit K deficiency (FII, VII , IX and X )
4. Congenital deficiency of factors
involved in extrinsic pathway.
5. DIC
6. inhibitors
43.
Partial Thromboplastin Time
(PTT)
Measures Effectiveness of the
Intrinsic Pathway(FVIII, FIX, FXI,
FXII, FII, FV, X).
It is the test Used for patients
receiving heparin therapy
NORMAL VALUE
25-40 SECS
Causes of prolongedPTT
Heparin therapy
Deficiency of factors involved in
intrinsic pathway, usually congenital:
Hemophilia A Hemophilia B and von
Willebrand disease)
DIC
Massive transfusion (labile FV, FVIII)
Inhibitors
46.
THROMBIN TIME (TT)
Timefor Thrombin To Convert
Fibrinogen Fibrin
A Measure of Fibrinolytic
Pathway
NORMAL VALUE
9-13 SECS
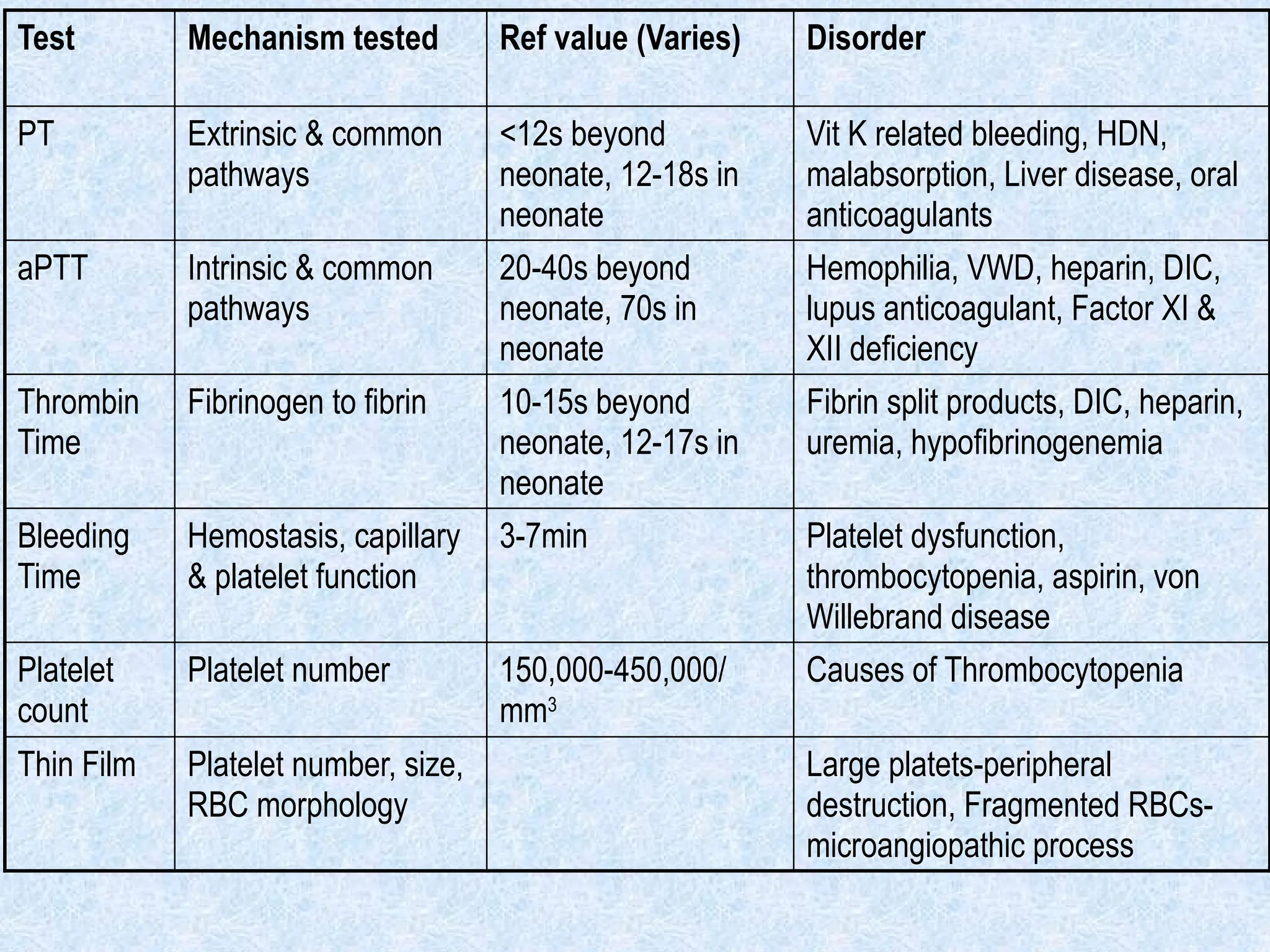
Test Mechanism testedRef value (Varies) Disorder
PT Extrinsic & common
pathways
<12s beyond
neonate, 12-18s in
neonate
Vit K related bleeding, HDN,
malabsorption, Liver disease, oral
anticoagulants
aPTT Intrinsic & common
pathways
20-40s beyond
neonate, 70s in
neonate
Hemophilia, VWD, heparin, DIC,
lupus anticoagulant, Factor XI &
XII deficiency
Thrombin
Time
Fibrinogen to fibrin 10-15s beyond
neonate, 12-17s in
neonate
Fibrin split products, DIC, heparin,
uremia, hypofibrinogenemia
Bleeding
Time
Hemostasis, capillary
& platelet function
3-7min Platelet dysfunction,
thrombocytopenia, aspirin, von
Willebrand disease
Platelet
count
Platelet number 150,000-450,000/
mm3
Causes of Thrombocytopenia
Thin Film Platelet number, size,
RBC morphology
Large platets-peripheral
destruction, Fragmented RBCs-
microangiopathic process
49.
PT and aPTT
•Prolonged APTT Defect in Intrinsic
No change in PT
• No change in APTT Defect in Extrinsic
Prolonged PT
• Prolonged APTT Defect in common
Prolonged PT
50.
Additional labs
Specificfactor assays
Mixing study (patient plasma 1:1 normal plasma)
Fibrinogen measurement
Platelet function testing
VWF antigen and activity testing
Genetic testing
51.
Coagulation factor disorders
•Inherited bleeding disorders
– Hemophilia A
– Hemophilia B
– vonWillebrands disease
– Other factor deficiencies
• Acquired bleeding disorders
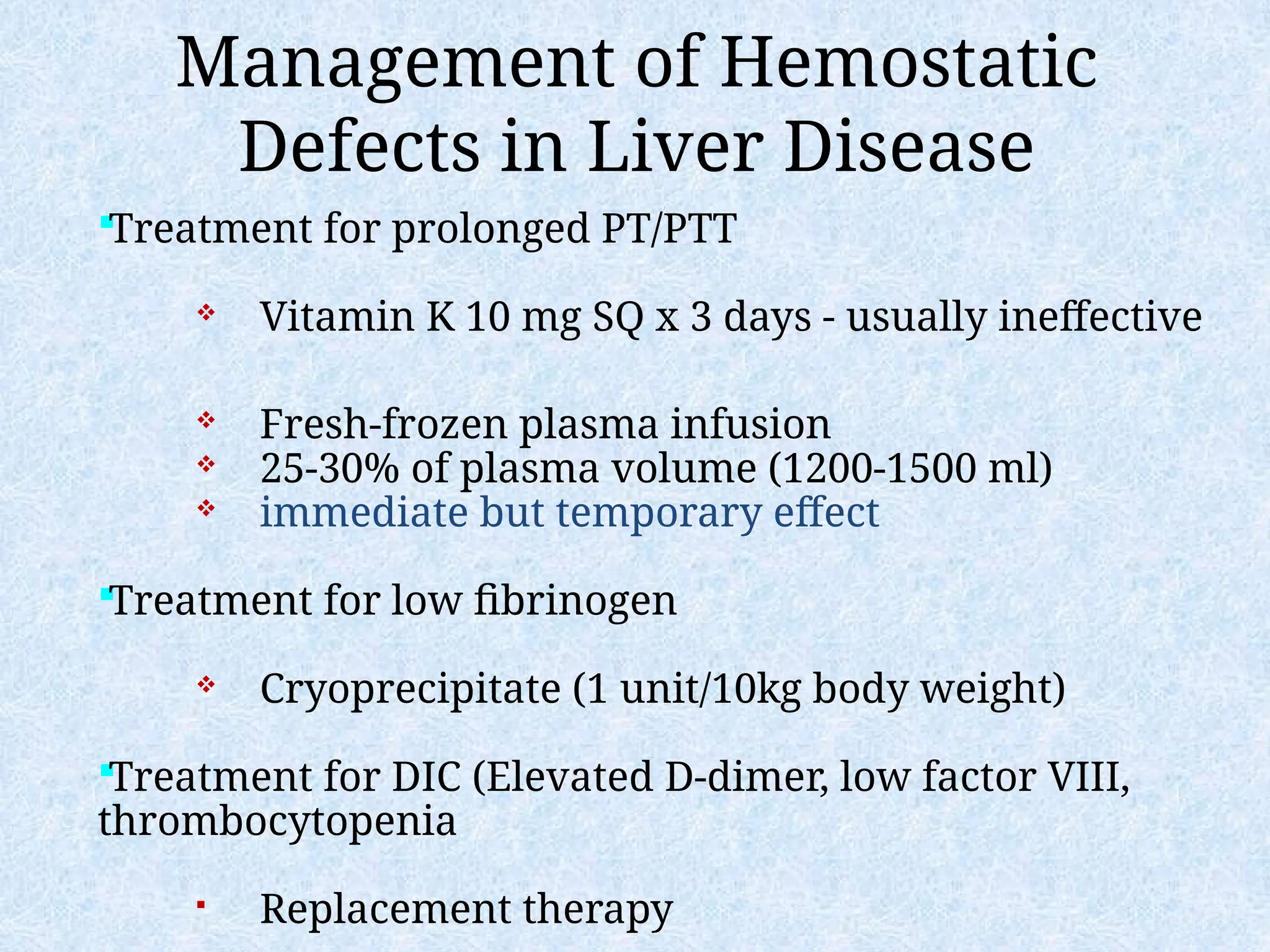
– Liver disease
– Vitamin K deficiency/warfarin overdose
– DIC
Factor VIII Deficiency
•Classic hemophilia (hemophilia A)
• X-linked disorder (affects 1º males)
• Most common - severe bleeding
• Spontaneous hematomas
• Prolonged PTT – Intrinsic path.
• Diagnosis - factor VIII assay
• Treatment:
•factor VIII concentrate
•Desmopressin
•Cryoprecipitate (less desirable)
54.
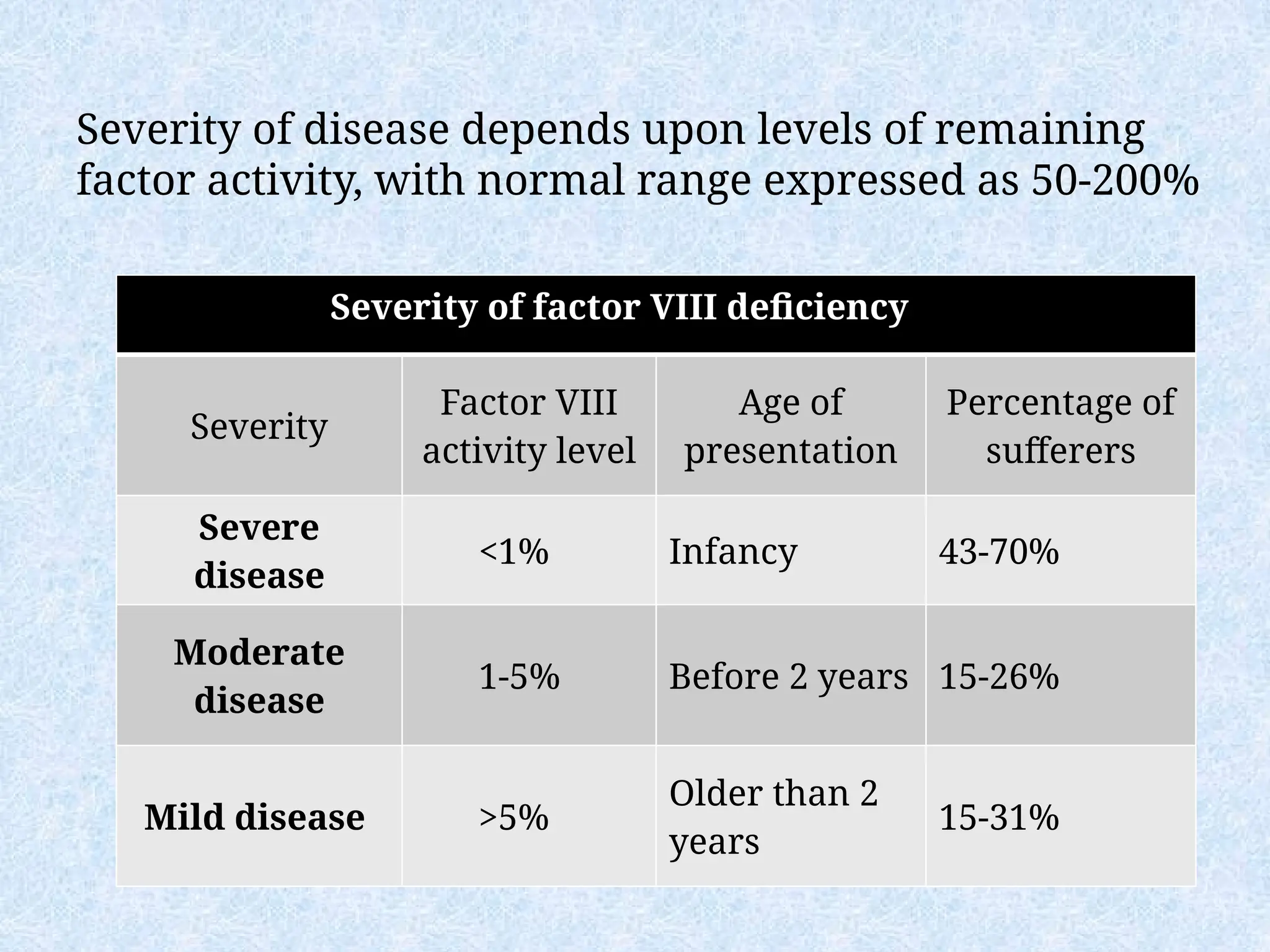
Severity of diseasedepends upon levels of remaining
factor activity, with normal range expressed as 50-200%
Severity of factor VIII deficiency
Severity
Factor VIII
activity level
Age of
presentation
Percentage of
sufferers
Severe
disease
<1% Infancy 43-70%
Moderate
disease
1-5% Before 2 years 15-26%
Mild disease >5%
Older than 2
years
15-31%
55.
Factor IX Deficiency
•Christmas disease (Hemophilia B):
•X-linked recessive disorder
•Indistinguishable from classic hemophilia (F VIII)
•Requires evaluation of factor VIII and IX activity levels to
diagnose
•Treatment:
•factor IX concentrate
•Cryoprecipitate if factor IX unavailable
•Desmopressin is not working
56.
Factor Deficiencies
Hemophilia A(Classic Hemophilia)
80-85% of all Hemophiliacs
Deficiency of Factor VIII
Lab Results - Prolonged PTT
HEMOPHILIA B (Christmas Disease)
10-15% of all Hemophiliacs
Deficiency of Factor IX
Lab Test - Prolonged PTT
INHIBITORS
30% of peoplewith haemophilia develop an antibody to
the clotting factor they are receiving for treatment.
These antibodies are known as inhibitors.
These patients are treated with high does of FVIIa for
bleeds or surgery. This overrides defect in FVIII or FIX
deficiency.
Longterm management involves attempting to eradicate
inhibitors by administering high dose FVIII (or FIX) in
a process called immune tolerance
60.
Dosing guidelines forhemophilia
A
• General measures: avoid trauma, traumatic sports and IM
injections
• Local measures: local pressure, ICE application
• Surgical: Joint replacement in case of joint deformity
• Factor VIII concentrate:
• Major surgery
– Target: 80-100% q8h; 7-14 days (or until wound healing)
– Dose: Desired level/2 x BW (kg)
• Minor surgery
– Target: 30-60% q12h; 3-5 days (or until wound healing)
– Dose: Desired level/2 x BW (kg)
• Prophylaxis:
– Dose is the same as in minor surgery but twice weekly
• Adjunctive therapy
– aminocaproic acid or DDAVP (for mild – mod disease
61.
Treatment of hemophiliaB
• Dose
–Dose: Desired level x BW (kg)
–The frequency in major and Minor
surgery as in Factor VIII
–Prophylaxis = 50 IU/kg once weekly
–No role for DDAVP in hemophilia B
62.
This is themost common
hereditary coagulopathy
in humans. It can be
congenital or acquired.
Pathophysiology
• Von Willebrand's
disease (vWD) results
from the deficiency or
abnormal function of
von Willebrand factor
(vWF).
• vWF is a multimeric
glycoprotein encoded for
by gene map locus 12p13.
• It is made in the
endothelium and stored
in Weibel-Palade bodies.
It has two main functions:
• It assists in platelet plug
formation by attracting
circulating platelets to the
site of damage.
• It binds to coagulation
factor VIII preventing its
clearance from the
plasma.
Von Willebrand's Disease
63.
Von Willebrand
factor
Von Willebrand
factoris a blood
glycoprotein
involved in
hemostasis. It is
deficient or defective
in von Willebrand
disease and is
involved in a large
number of other
diseases, including
thrombotic including
thrombotic thrombocyto
penic
purpura, Heyde's
syndrome, and possibly
64.
Etiology
I. Hereditary -three types
• vWD Type I, vWD Type II, and vWD Type III
• Within the three inherited types of vWD there are various
subtypes.
II. Acquired - also called pseudo-von Willebrand's disease
or platelet-type; it is frequently found in:
• Lymphoproliferative
• Myeloproliferative disorders
• Solid tumors
• Immunological disorders
• Cardiovascular disorders e.g., aortic stenosis,
• Wilms'tumor,
• Hypothyroidism.
Von Willebrand's Disease
65.
Laboratory evaluation of
vonWillebrand disease
• Classification
– Type 1 Partial quantitative deficiency
– Type 2 Qualitative deficiency
– Type 3 Total quantitative deficiency
• Diagnostic tests:
vonWillebrand type
Assay 1 2 3
vWF antigen ß Normal ßß
vWF activity ß ß ßß
Multimer analysis Normal Normal
Absent
66.
Types of hereditaryvon Willebrand's disease (vWD)
Type 1 60-80% Quantitative
defect (19-45%
of enzyme level
present)
• Heterozygous
for defective
gene
• Inherited as AD
• Normal lifespan
• Occasionally easy
bruising and/or
menorrhagia
• Bleeding after
dental work,
major surgery
Type 2 20-30% Qualitative
defect -
multimers
abnormal or
subgroups
absent
Usually AD
inheritance
(rarely AR)
Bleeding tendency
varies
Four subtypes:
2A, 2B, 2M, 2N
Type 3 Rare - the
most
severe
form; 1-
Quantitative -
levels very low
or undetectable
• Homozygous
for defective
gene
• AR inheritance
• Severe mucosal
bleeding
• May have
haemarthrosis (as
Von Willebrand's Disease
67.
Treatment of vonWillebrand
Disease
• Cryoprecipitate
– Source of fibrinogen, factor VIII and VWF
– Only plasma fraction that consistently contains VWF
multimers
– 1 unit / 10 kg
• DDAVP (deamino-8-arginine vasopressin)
– plasma VWF levels by stimulating secretion from
endothelium
– Duration of response is variable
– Not generally used in type 2 disease
– Dosage 0.3 µg/kg q 12 hr IV
• Factor VIII concentrate (Intermediate purity)
– Virally inactivated product
68.
Vitamin K deficiency
•Source of vitamin K Green vegetables
Synthesized by intestinal
flora
• Required for synthesis Factors II, VII, IX ,X
Protein C and S
• Causes of deficiency Malnutrition
Biliary obstruction
Malabsorption
Antibiotic therapy
• Treatment Vitamin K
Fresh frozen plasma
69.
Common clinical conditionsassociated with
Disseminated Intravascular Coagulation
• Sepsis
• Trauma
– Head injury
– Fat embolism
• Malignancy
• Obstetrical
complications
– Amniotic fluid embolism
– Abruptio placentae
• Vascular disorders
• Reaction to toxin (e.g.
snake venom, drugs)
• Immunologic disorders
– Severe allergic reaction
– Transplant rejection
Activation of both coagulation and fibrinolysis
Triggered by
70.
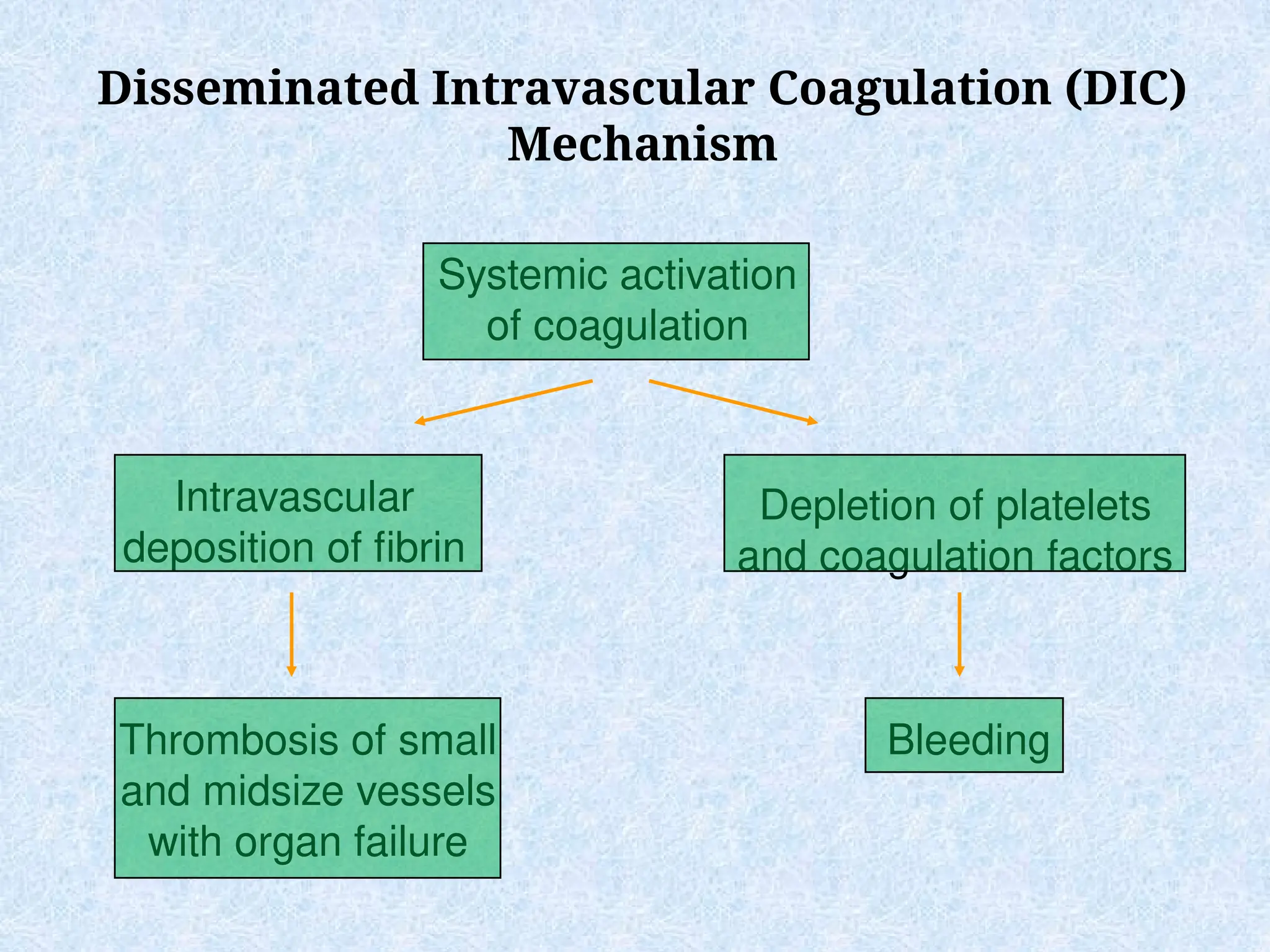
Disseminated Intravascular Coagulation(DIC)
Mechanism
Systemic activation
of coagulation
Intravascular
deposition of fibrin
Depletion of platelets
and coagulation factors
Bleeding
Thrombosis of small
and midsize vessels
with organ failure
71.
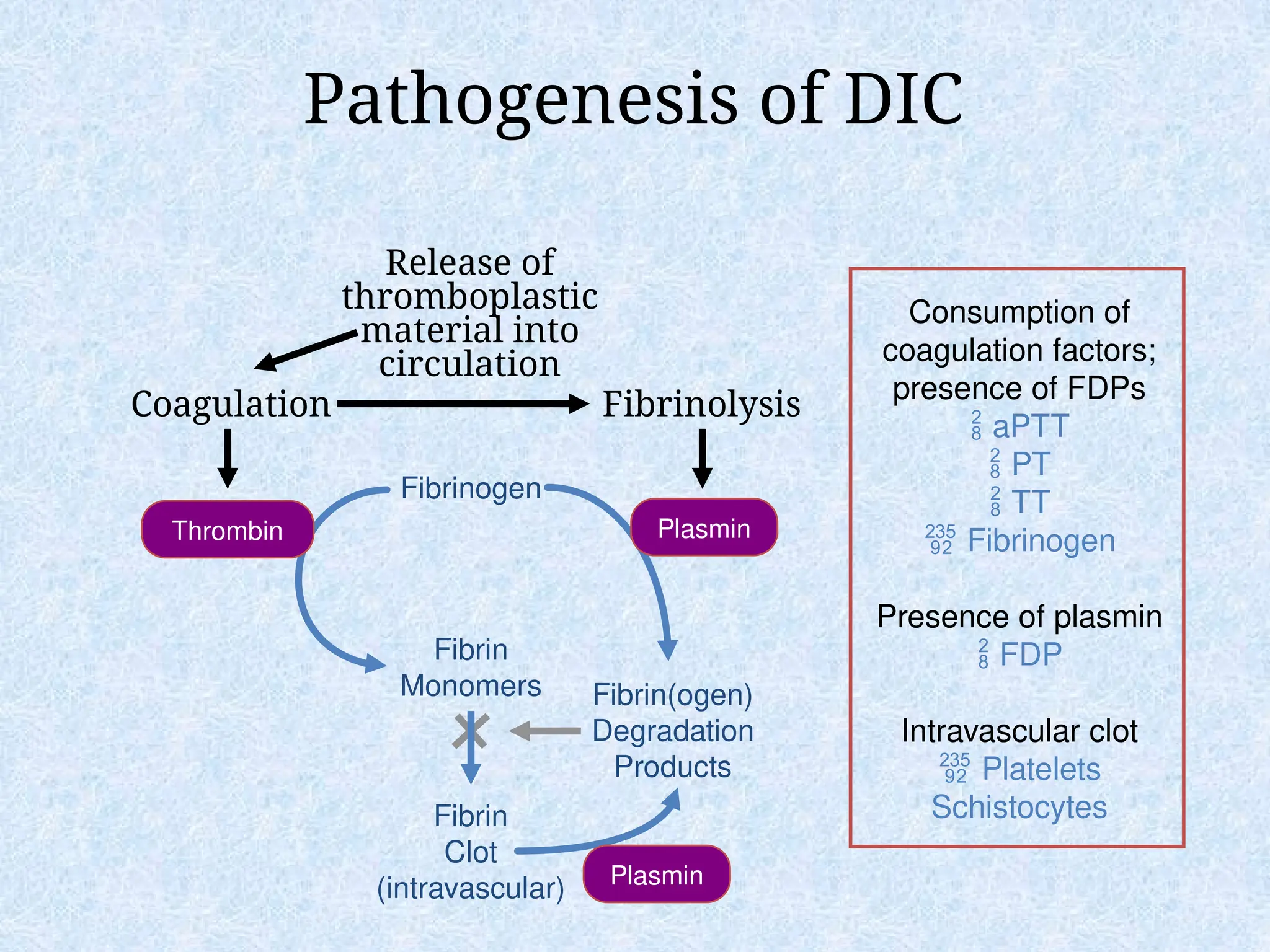
Pathogenesis of DIC
CoagulationFibrinolysis
Fibrinogen
Fibrin
Monomers
Fibrin
Clot
(intravascular)
Fibrin(ogen)
Degradation
Products
Plasmin
Thrombin Plasmin
Release of
thromboplastic
material into
circulation
Consumption of
coagulation factors;
presence of FDPs
aPTT
PT
TT
Fibrinogen
Presence of plasmin
FDP
Intravascular clot
Platelets
Schistocytes
Cryoprecipitate
• Prepared fromFFP
• Content
–Factor VIII, von Willebrand factor,
fibrinogen
• Indications
–Fibrinogen deficiency
–Uremia
–von Willebrand disease
• Dose (1 unit = 1 bag)
–1-2 units/10 kg body weight
82.
Hemostatic drugs
Aminocaproic acid(Amicar)
• Mechanism
– Prevent activation plaminogen -> plasmin
• Dose
– 50mg/kg po or IV q 4 hr
• Uses
– Primary menorrhagia
– Oral bleeding
– Bleeding in patients with thrombocytopenia
– Blood loss during cardiac surgery
• Side effects
– GI toxicity
– Thrombi formation
83.
Hemostatic drugs
Desmopressin (DDAVP)
•Mechanism
– Increased release of VWF from endothelium
• Dose
– 0.3µg/kg IV q12 hrs
– 150mg intranasal q12hrs
• Uses
– Most patients with von Willebrand disease
– Mild hemophilia A
• Side effects
– Facial flushing and headache
– Water retention and hyponatremia
84.
Recombinant human factorVIIa
(rhVIIa;
• Mechanism
– Direct activation of common pathway
• Use
– Factor VIII inhibitors
– Bleeding with other clotting disorders
– Warfarin overdose with bleeding
– CNS bleeding with or without warfarin
– Dose
– 90 µg/kg IV q 2 hr
– “Adjust as clinically indicated”
85.
Approach to bleedingdisorders
Summary
• Identify and correct any specific defect of
hemostasis
– Laboratory testing is almost always needed to establish
the cause of bleeding
– Screening tests (PT,PTT, platelet count) will often allow
placement into one of the broad categories
– Specialized testing is usually necessary to establish a
specific diagnosis
• Use non-transfusional drugs whenever
possible
• RBC transfusions for surgical procedures or
large blood loss




















![Prothrombin Time (PT)
Measures The Effectiveness of the
Extrinsic Pathway (FVII, FII, FV, X)
[majority are vitamin K dependent
factor].
Used for monitoring oral anticoagulant
therapy and used as a liver function
test
Normal range: 10-14 sec](https://image.slidesharecdn.com/00-approachofbleedingtendencyinadults-251003142320-a60ad3f0/75/00-Approach-of-bleeding-Tendency-in-Adults-ppt-40-2048.jpg)