Download to read offline

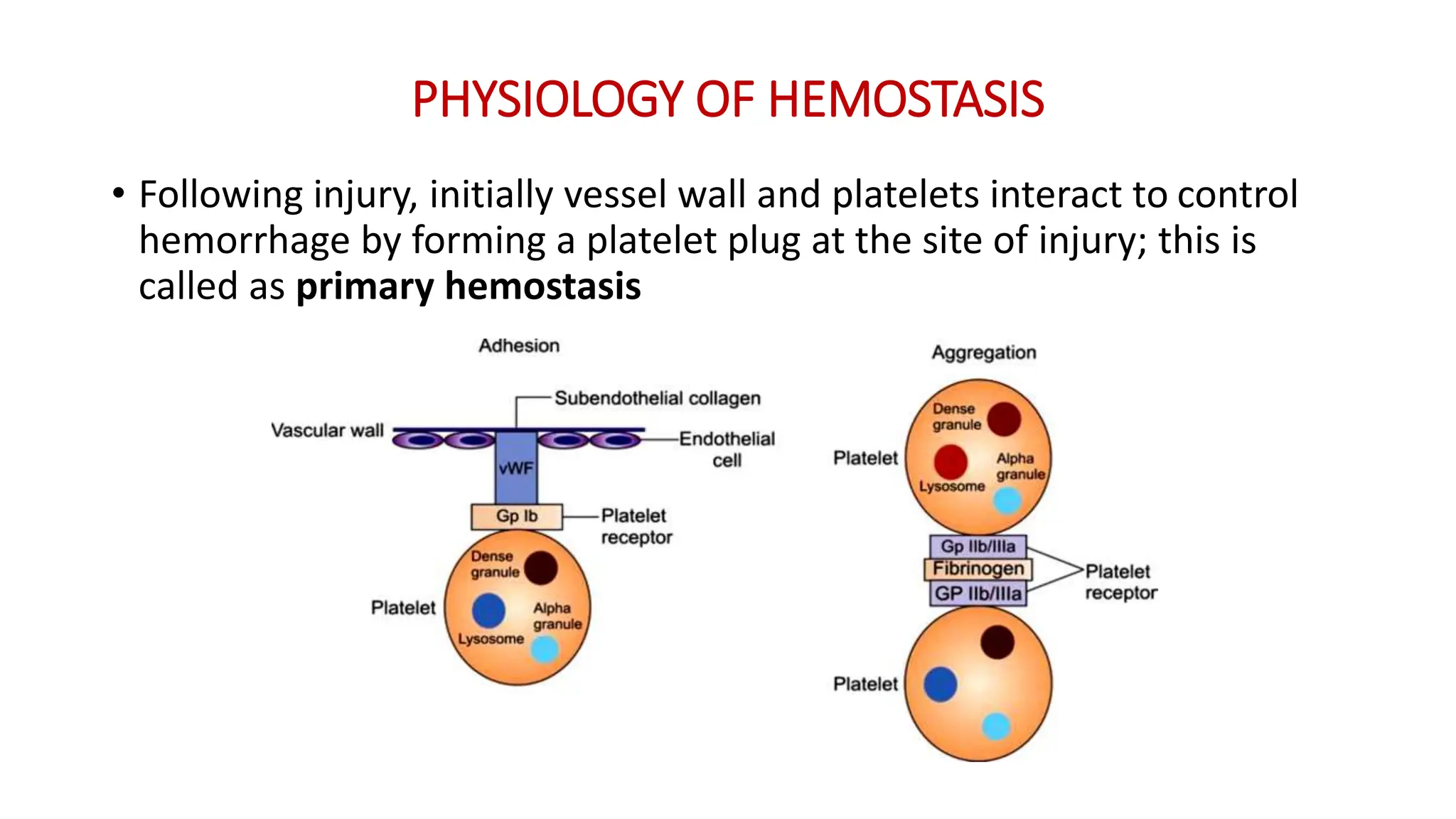


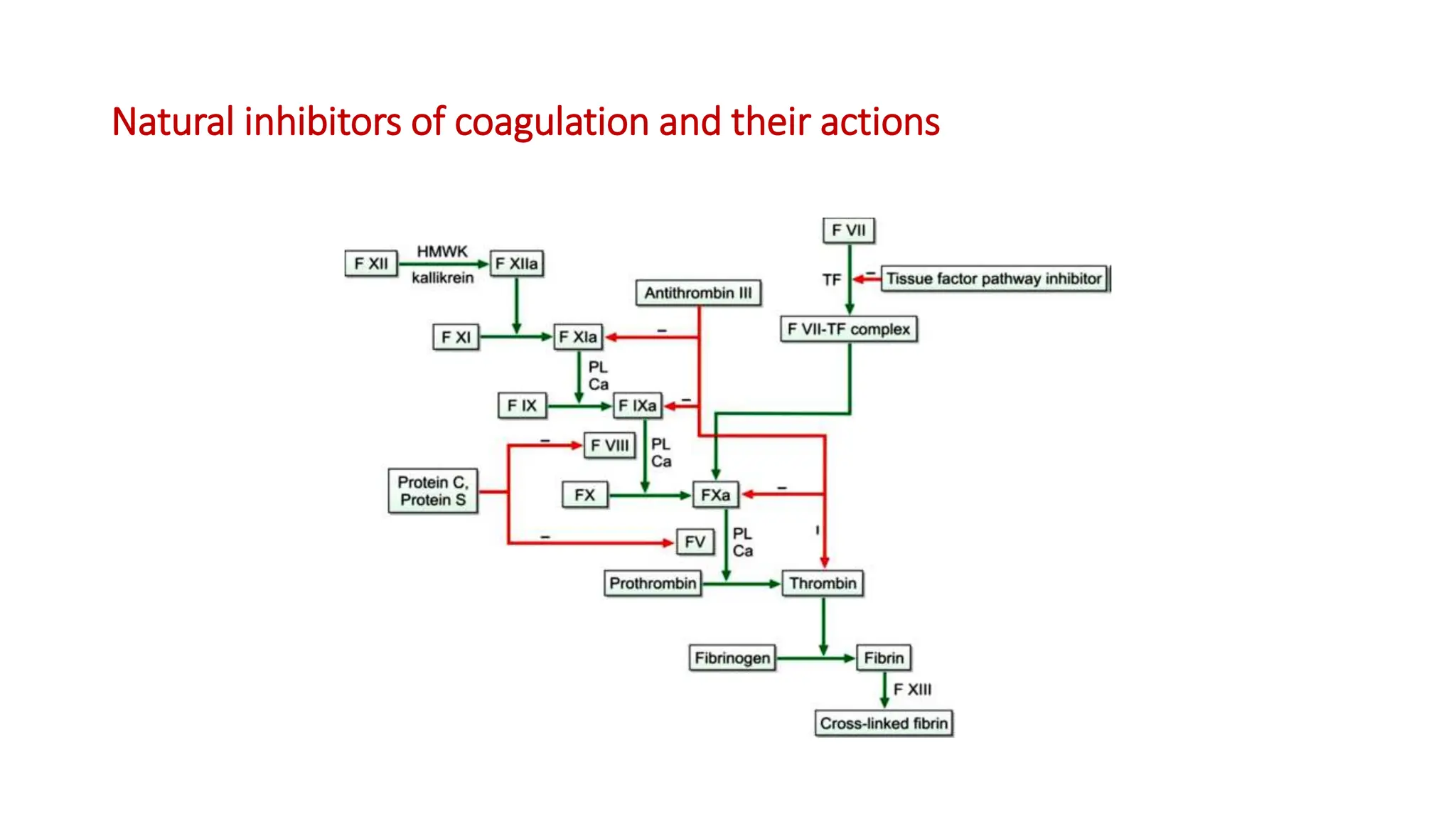
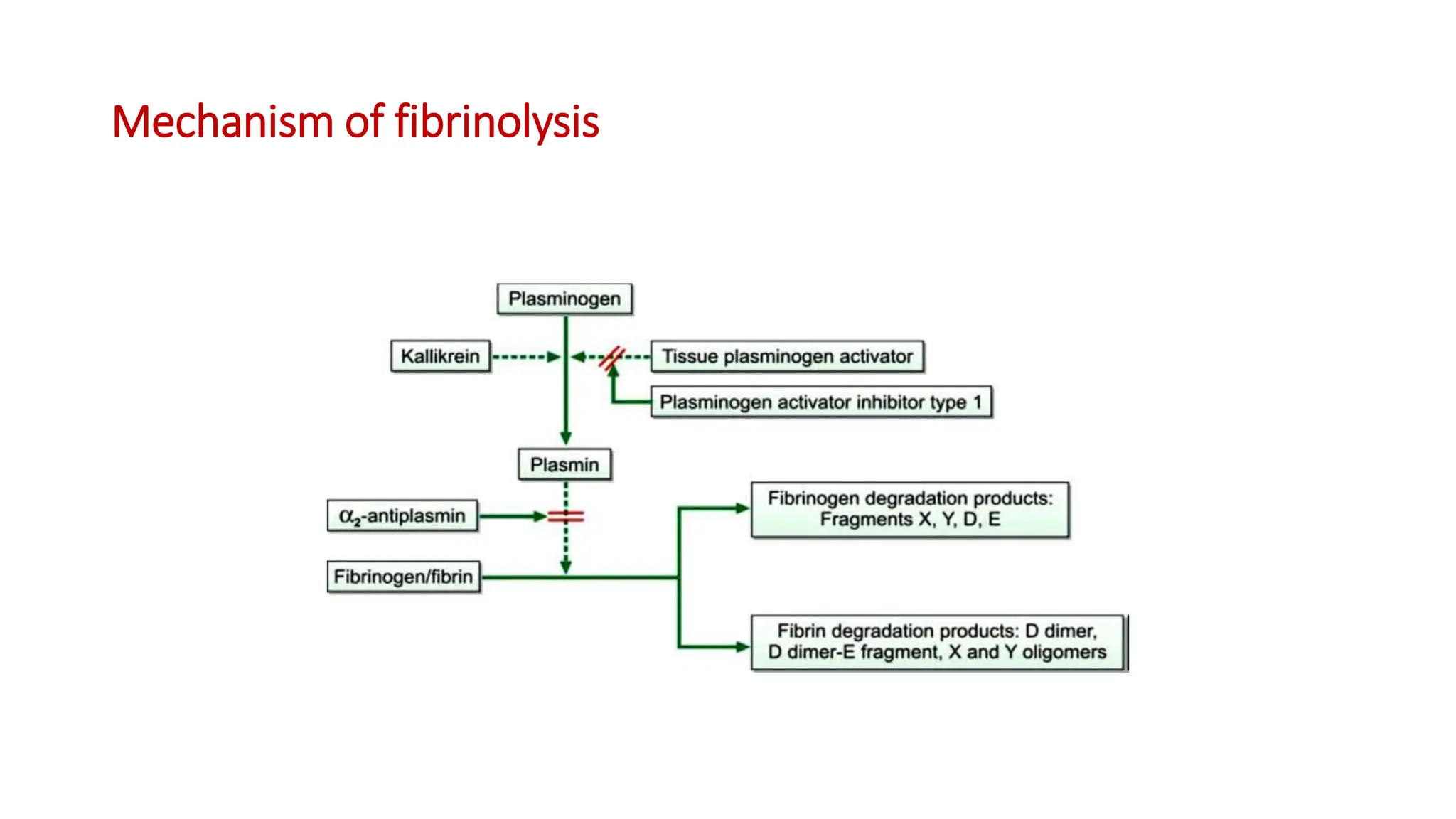










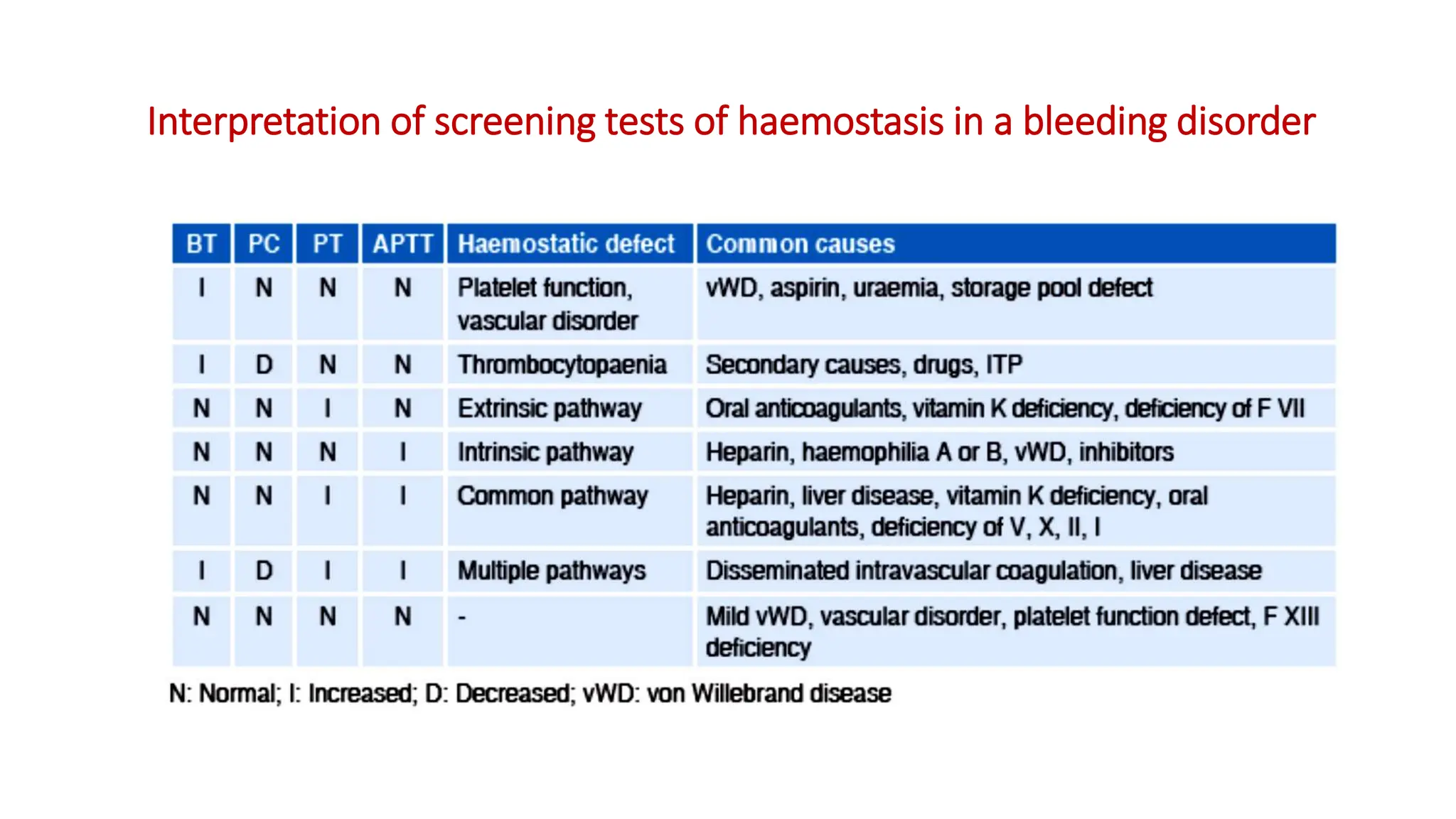
































Bleeding disorders result from defects in hemostasis due to abnormalities in blood vessels, platelets, or coagulation factors. Hemostasis involves primary hemostasis where platelets form a platelet plug and secondary hemostasis where a fibrin clot is formed. Common bleeding disorders include hemophilia A which is a coagulation factor VIII deficiency mostly affecting males, immune thrombocytopenia where autoantibodies destroy platelets, and von Willebrand disease where von Willebrand factor is defective. Screening tests for bleeding disorders include bleeding time, prothrombin time, activated partial thromboplastin time, and thrombin time which assess platelet function and levels of coagulation factors.


























































