Downloaded 63 times



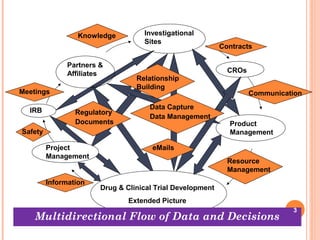

![WHY DO WE NEED A DATA
MANAGEMENT & DATA INTEGRITY
SYSTEM?
Enormous volumes of data
Example, a Phase-III trial in 10 centres with
100 patients each
60 pages of CRF for each recruited patient
20 fields each page
40 pages of screening form for each candidate
patient
20 fields each page
[1000 (60 x 20)] + [1500 (40 x 20)]
= 12, 00000 + 12, 00000
= 24,00000 specific data points 5](https://image.slidesharecdn.com/developingprotocolsproceduresforctdataintegrityfinalfinal-170302043711/85/Developing-Protocols-Procedures-for-CT-Data-Integrity-5-320.jpg)






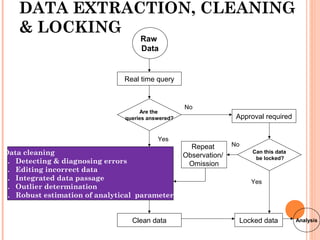





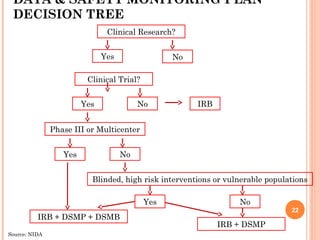











The document outlines protocols and procedures for ensuring clinical trial data integrity, emphasizing the importance of accurate data management and monitoring to maintain research integrity. It discusses various monitoring techniques, including risk-based monitoring, and the roles of data and safety monitoring plans (DSMP) and boards (DSMB) in safeguarding trial subjects and ensuring the validity of study outcomes. The document concludes by highlighting the critical need for high-quality data to uphold the credibility of clinical research and regulatory compliance.











































































































