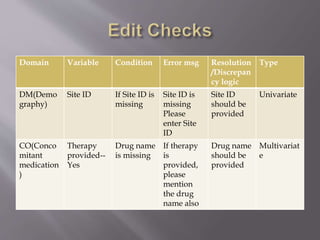
Clinical data management is the process of collecting, validating, and cleaning data from clinical trials. It aims to ensure data quality and integrity. Key aspects of clinical data management include electronic data capture, establishing data standards, using clinical data management systems, and performing activities like data collection, validation, and discrepancy management. It follows guidelines from organizations like SCDM and regulations like 21 CFR Part 11.