Downloaded 355 times











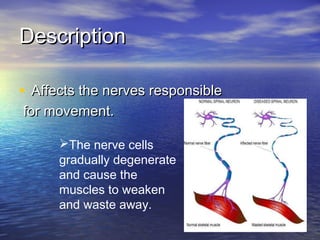




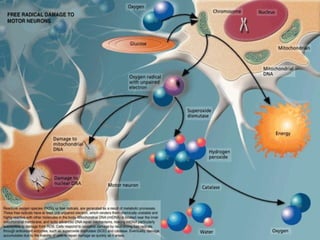





















This document discusses degenerative diseases and focuses on motor neuron disease (MND). It defines MND as the progressive degeneration of motor neurons in the spinal cord, brain stem, and motor cortex. The document covers the pathology, epidemiology, genetics, clinical features, investigations, management, and prognosis of MND. It describes different variants of MND including amyotrophic lateral sclerosis, primary lateral sclerosis, progressive muscular atrophy, and bulbar onset disease. No cure currently exists for MND, though riluzole has been shown to modestly prolong survival.



































































































