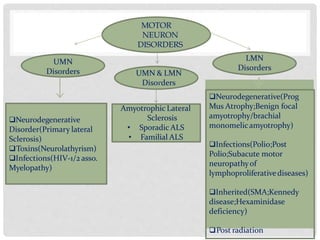
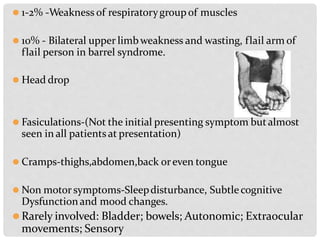
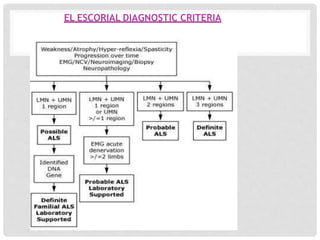
Motor neuron diseases are a group of neurodegenerative disorders that affect motor neurons in the brain and spinal cord. The presentation and progression of symptoms varies depending on whether upper motor neurons, lower motor neurons, or both are affected. Amyotrophic lateral sclerosis (ALS) is the most common type of motor neuron disease in adults and causes progressive weakness and atrophy as motor neurons degenerate. While there is no cure for motor neuron diseases, treatment focuses on managing symptoms and maximizing quality of life.