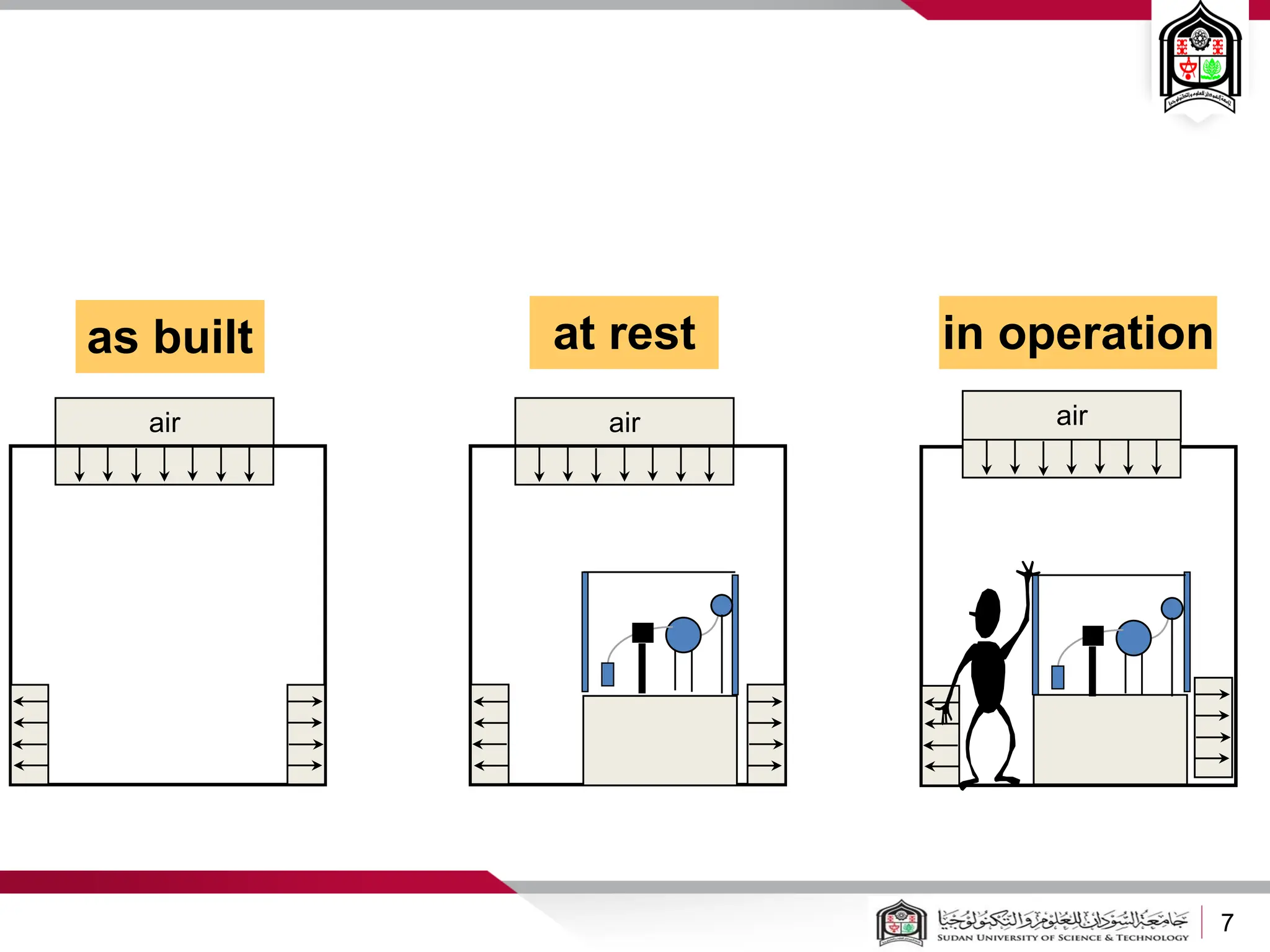
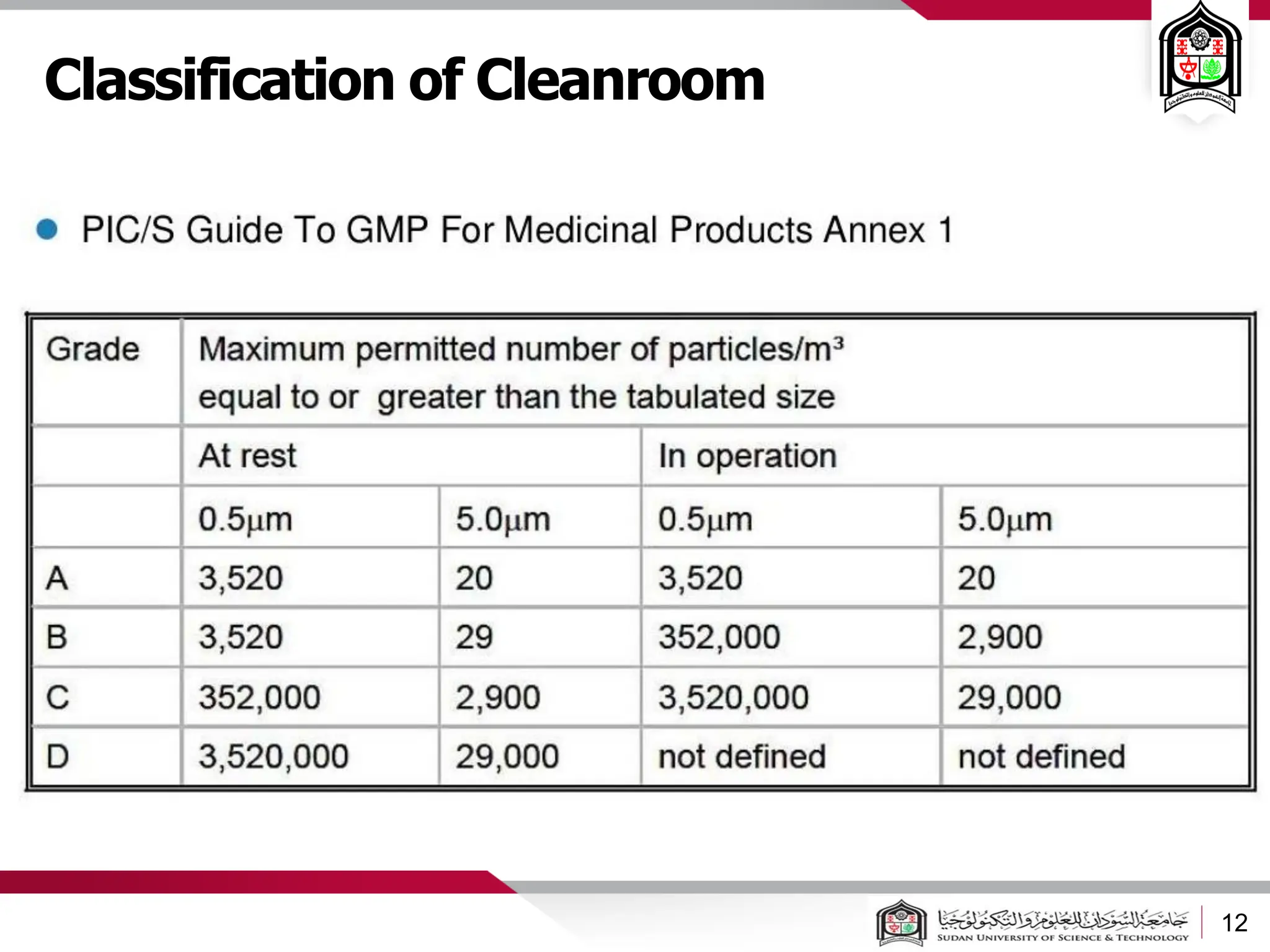
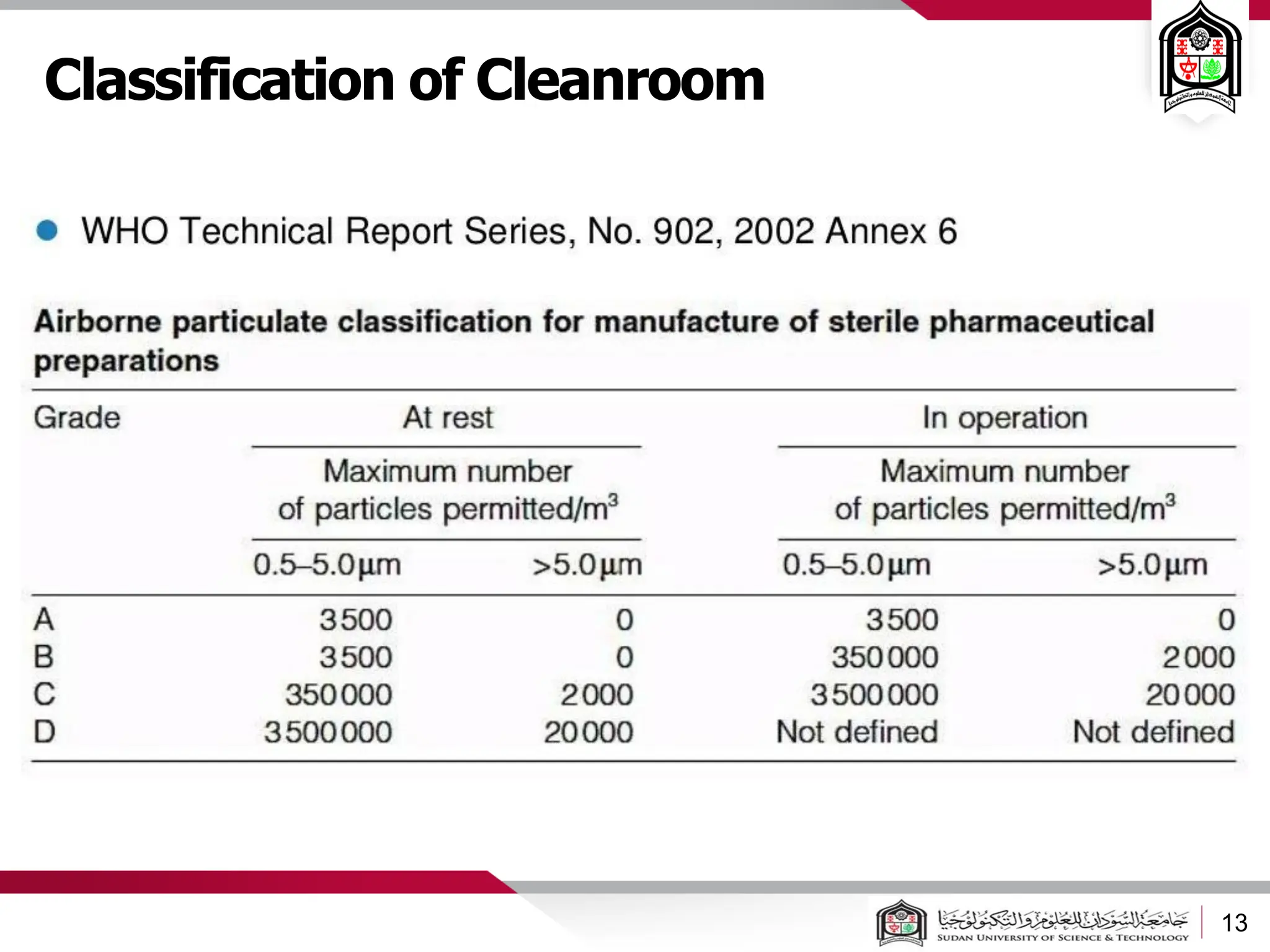
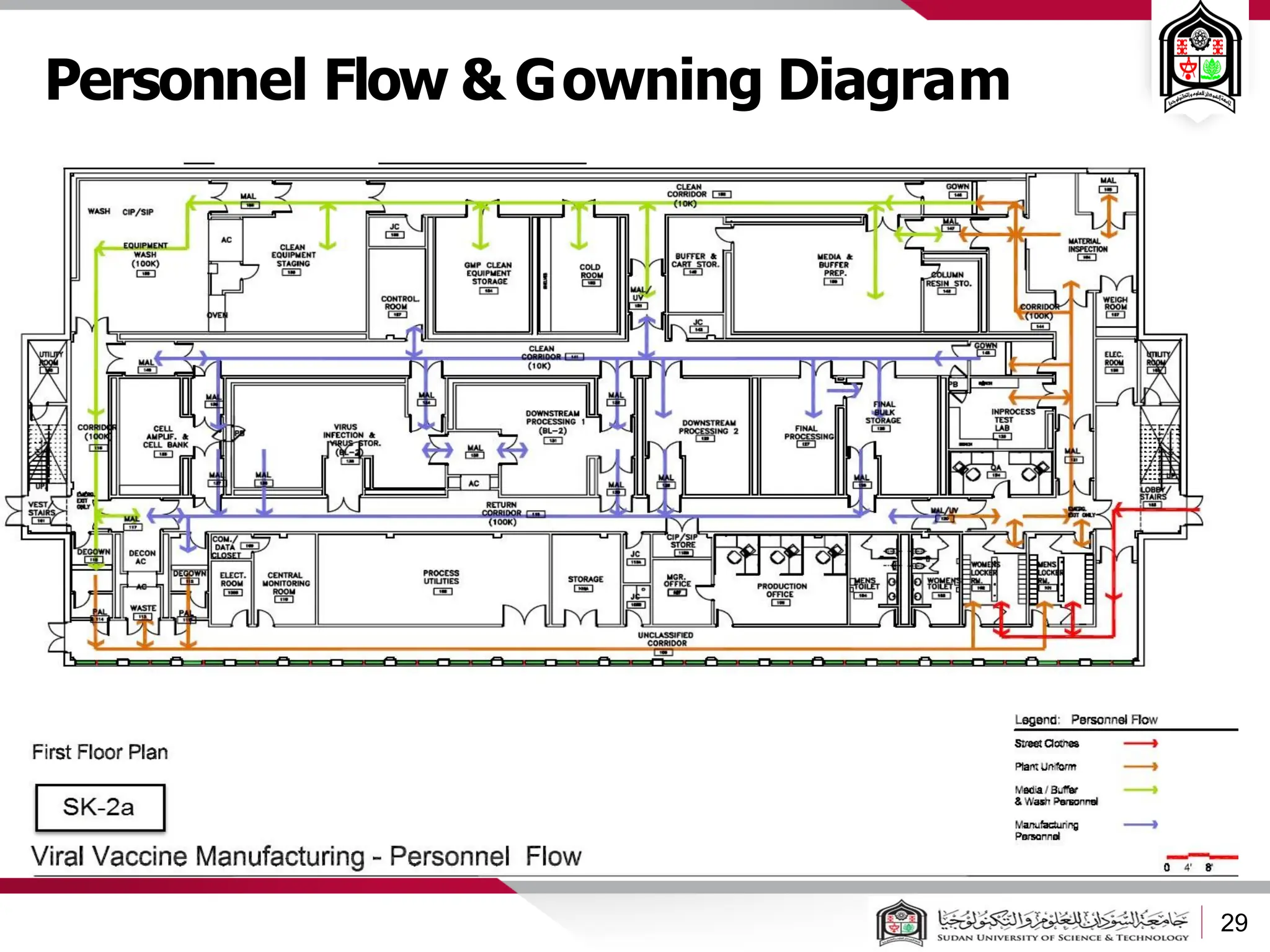
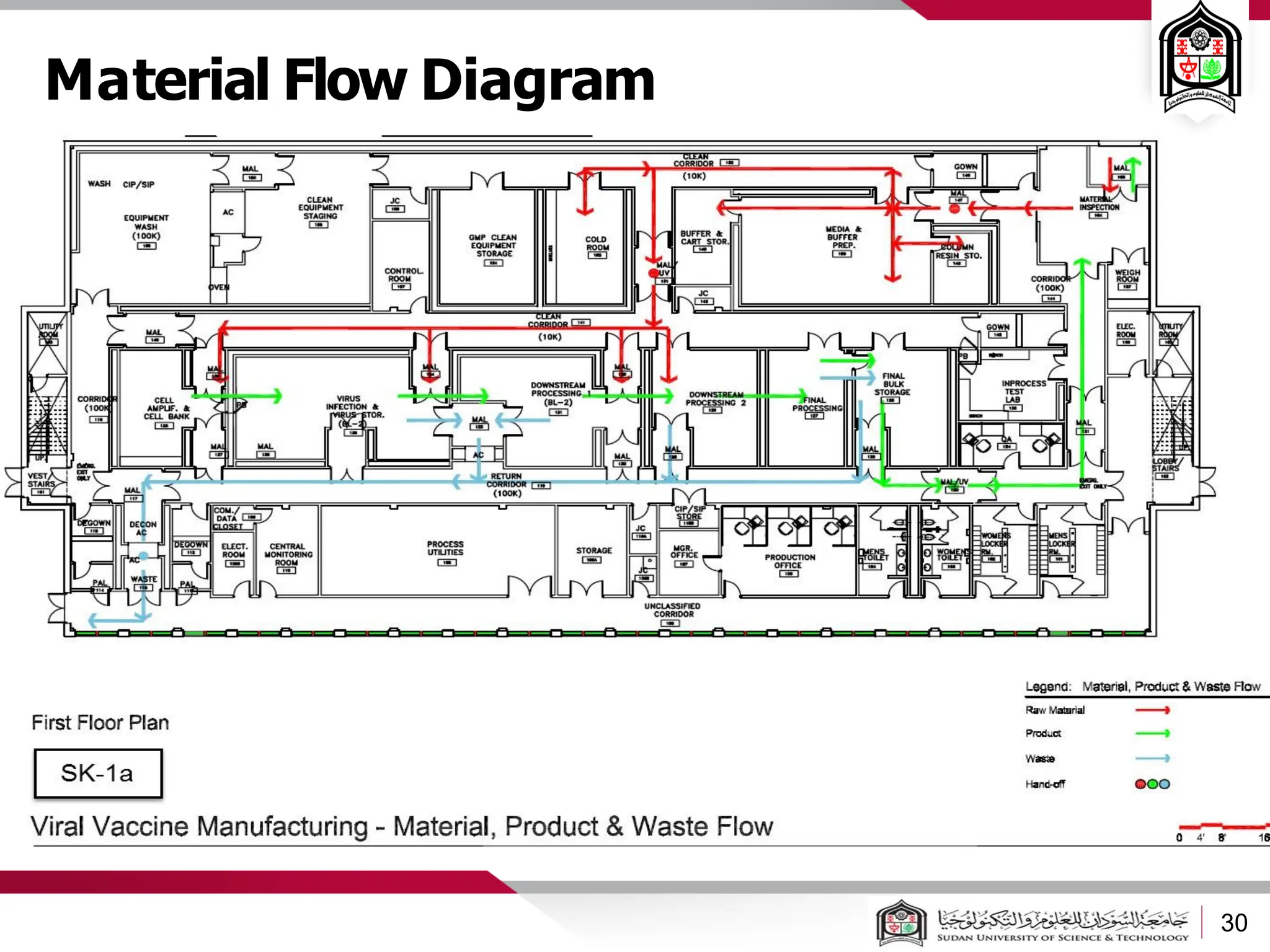
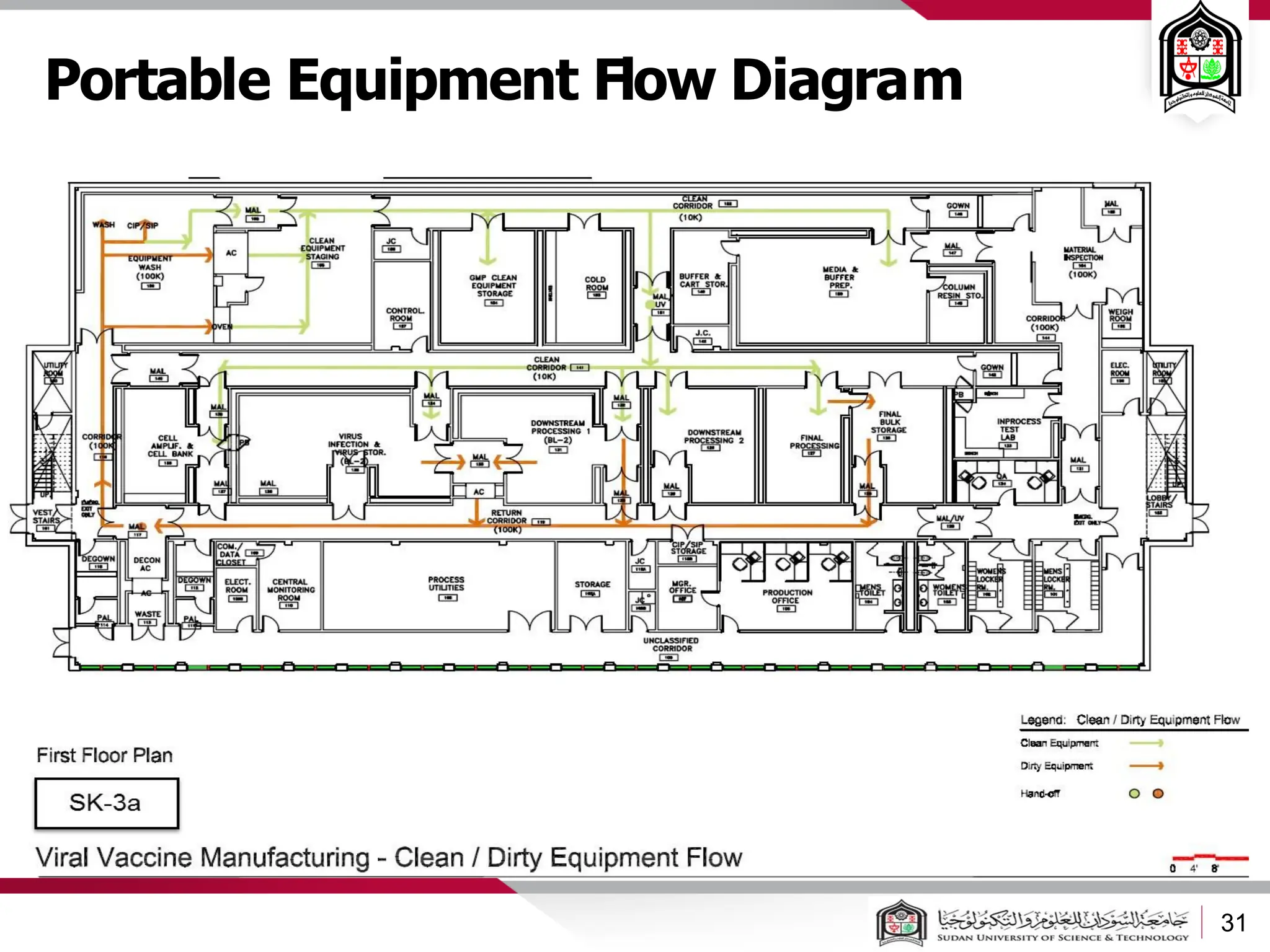
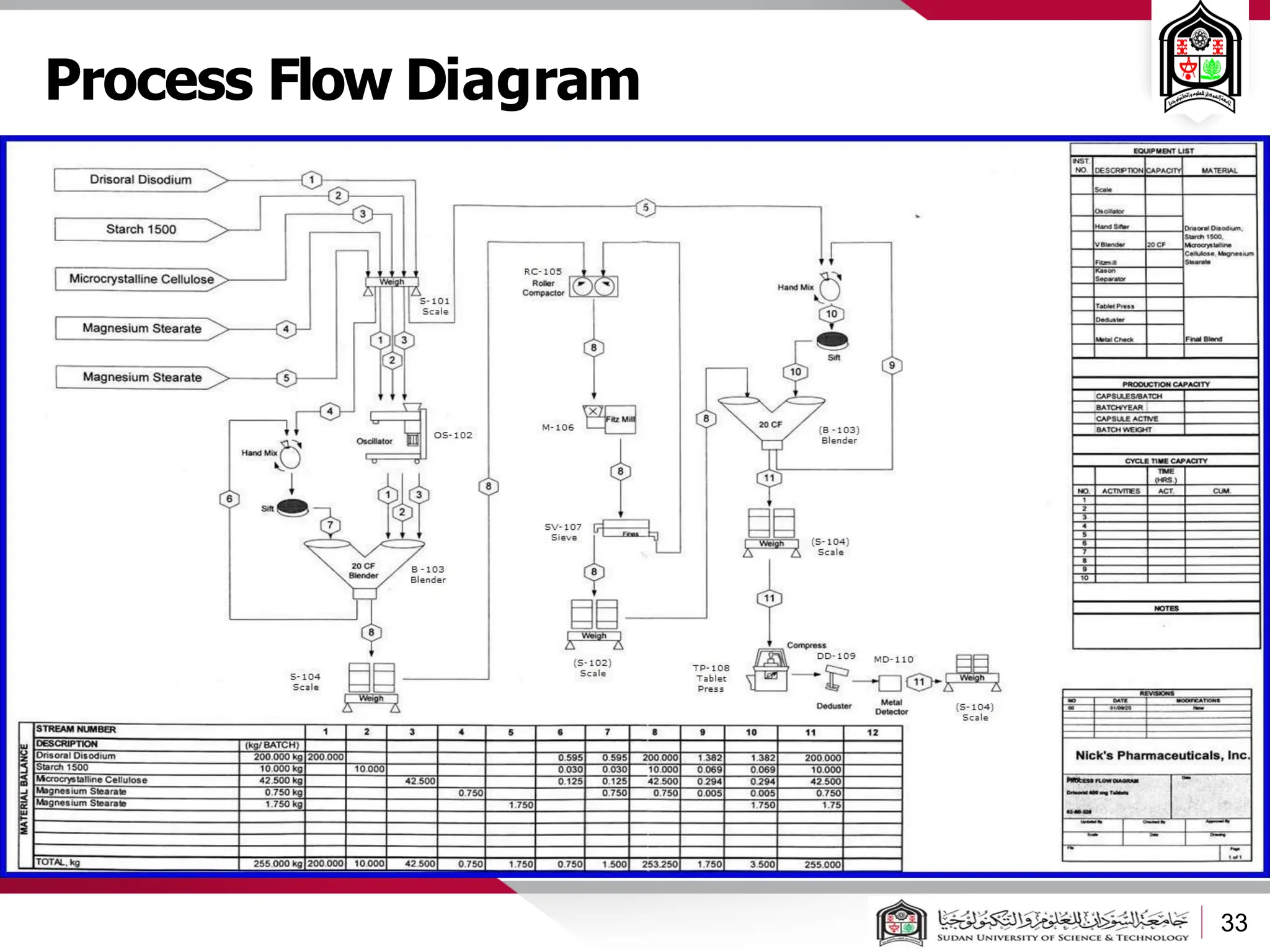
The document discusses cleanrooms and their classification systems according to ISO standards and PIC/S guidelines. It provides information on cleanroom design features and layout considerations for pharmaceutical manufacturing facilities. The key steps in sterile vial filling are outlined, and traditional cleanroom-based approaches are compared to newer isolator technologies that remove the operator from the sterile processing area.