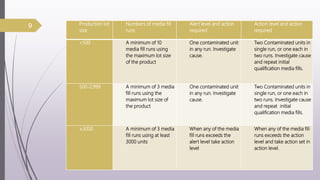
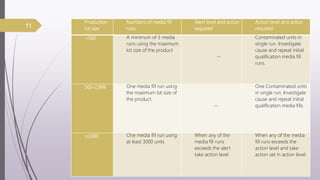
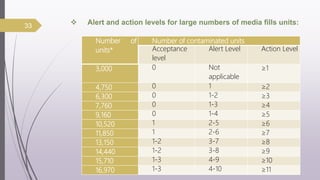
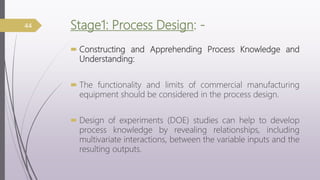
This document summarizes guidelines for media fill validation and USFDA process validation approaches. It discusses media fill validation, including why it is required, how to conduct media fill tests, parameters that affect sterility, and requalification requirements. It provides details on media fill procedures for liquid, powder, and lyophilized products. Frequency of media fills depends on production batch size, with more runs required for initial qualification and after certain changes. The document also introduces the USFDA's process validation lifecycle approach, which focuses on validating processes throughout the product lifecycle rather than just at startup.












































































































![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)










![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)





