Downloaded 322 times















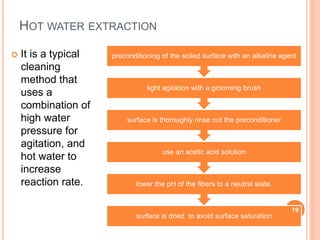















Cleaning validation is an important process in the pharmaceutical industry to ensure product safety and purity. It involves documenting evidence that an approved cleaning procedure will adequately clean equipment used in pharmaceutical production. The cleaning validation process includes planning, execution, analytical testing, and reporting phases. A cross-functional team plans the validation program, which involves grouping products, equipment, cleaning agents, and methods. Sampling techniques like swab and rinse sampling are used in the execution phase. Acceptance criteria are established and analytical tests are performed on samples to verify cleaning levels. A validation report documents the results and conclusions to obtain approval. Revalidation may be required if any changes are made to the cleaning process.























































































