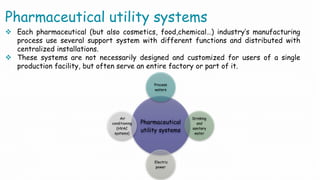
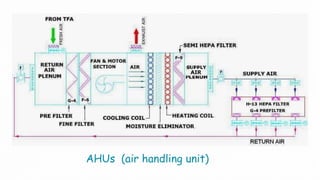
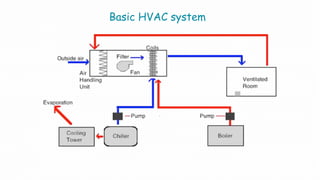
The document summarizes validation of an HVAC system for a pharmaceutical facility. It discusses the importance of HVAC systems in cleanrooms and outlines some key validation parameters to test, including:
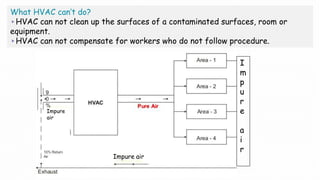
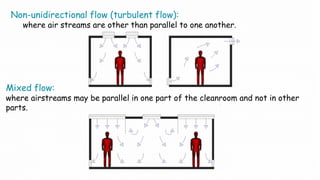
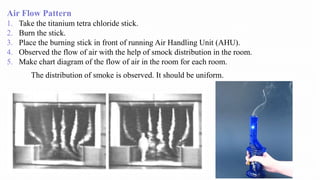
1. Airflow pattern, velocity, and changes per hour to ensure proper airflow.
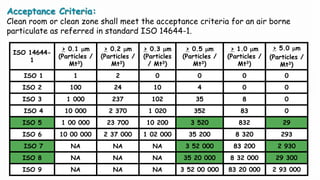
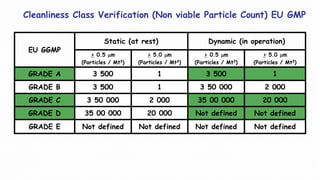
2. Filter leak testing and particulate counting to check filter performance and air quality.
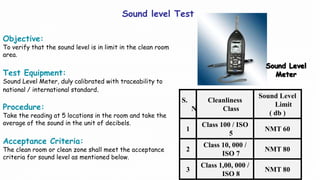
3. Pressure differential, temperature, humidity, and sound level testing to validate environmental controls.
Validation of the HVAC system is necessary to demonstrate that it can consistently supply air meeting quality standards to maintain aseptic manufacturing conditions.

































![• [Downloaded from http://www.asiapharmaceutics.info on Wednesday,
October 01, 2014, IP: 223.30.225.254] || Click here to download free
Android application for this journal
• http://www.gmpua.com/CleanRoom/HVAC/Solid.pdf
References
• Validation in pharmaceutical industry concepts, approaches &
guidelines by P.P.Sharma , 1st edition published by vandana
publications PVT.LTD dehli (page no. 165- 191)](https://image.slidesharecdn.com/validationofhvacsystem-190127131909/85/Validation-of-hvac-system-42-320.jpg)




































































