The document discusses validation of computerized systems used in the pharmaceutical industry. It outlines the importance of validation to ensure accuracy, reliability and consistent performance. The key aspects covered include validation protocols, qualification of hardware and software, user requirements, change control, and compliance with regulations like 21 CFR Part 11 and guidance like GAMP 5. The overall goal of validation is to confirm that computer systems meet intended uses and fulfill requirements.
“Confirmation by examinationand provision of objective
evidence that computer system specifications conform to user
needs and intended uses and that all requirements can be
consistently fulfilled”.
2
3.
Ensure anacceptable degree of documented evidence that
establishes confidence in the accuracy, reliability and
consistency in performance of the system.
Meet the principles ALCOA.
Ensure that all technical and procedural controls are
implemented ensuring compliance with good documentation
practices for electronic data generated by the system
3
VALIDATION
The computerized systemshall be validated in accordance with
the Corporate Standards and regulatory requirements to ensure:
Accuracy.
Reliability.
Consistent Intended Performance.
Ability to discern invalid or altered records.
Evidence of validation.
5
6.
CSVMP
Describes thepolicy, approach, organization and planning,
resources, execution and management of computerized
system validation for all of the GXP systems in use on-site.
Contain, the scope, risk management approach and a
complete inventory list of all GXP systems.
Validation should be executed in accordance with the
validation protocol and applicable SOPs.
6
7.
7
Validation reportoutlines the validation process and activities
and describe and justify any deviations from the process and
activities specified in the protocol.
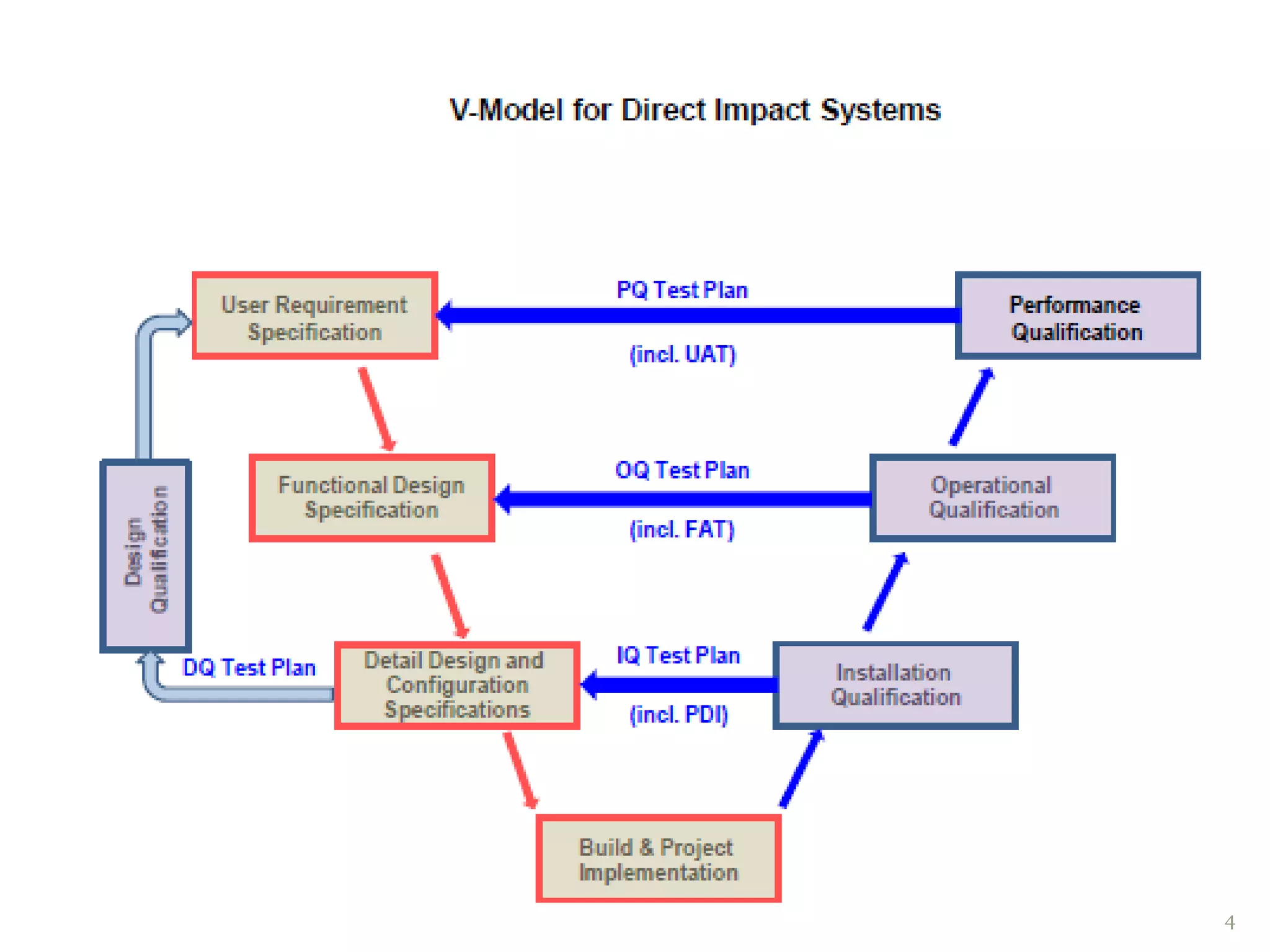
Design qualification .
Installation qualification.
Operational qualification.
:hardware qualification
:software qualification.
Standard operating procedures and training.
Performance qualification.
8.
HARDWARE QUALIFICATION
Thequalification of the hardware should prove:
capacity of the hardware matches its assigned function.
operates within the operational limits
hardware configuration settings are appropriate and meet
user and functional requirements;
performs acceptably under challenging conditions
reproducibility/consistency.
8
9.
SOFTWARE QUALIFICATION
Functionaltesting of software should provide assurance that
computer programs will function consistently within pre-
established limits for both normal conditions as well as under
worst-case conditions.
Functional testing, also known as “black box” testing,
involves inputting normal and abnormal test cases; then,
evaluating outputs against those expected.
9
10.
SYSTEM OPERATION ANDMAINTENANCE
Periodically reviewed to determine whether the system
remains in a validated state or whether there is a need for
revalidation.
The review should cover:
• review of changes;
• review of deviations;
• review of incidents;
• systems documentation;
• procedures;
• training;
• effectiveness of corrective and preventive action (CAPA). 10
11.
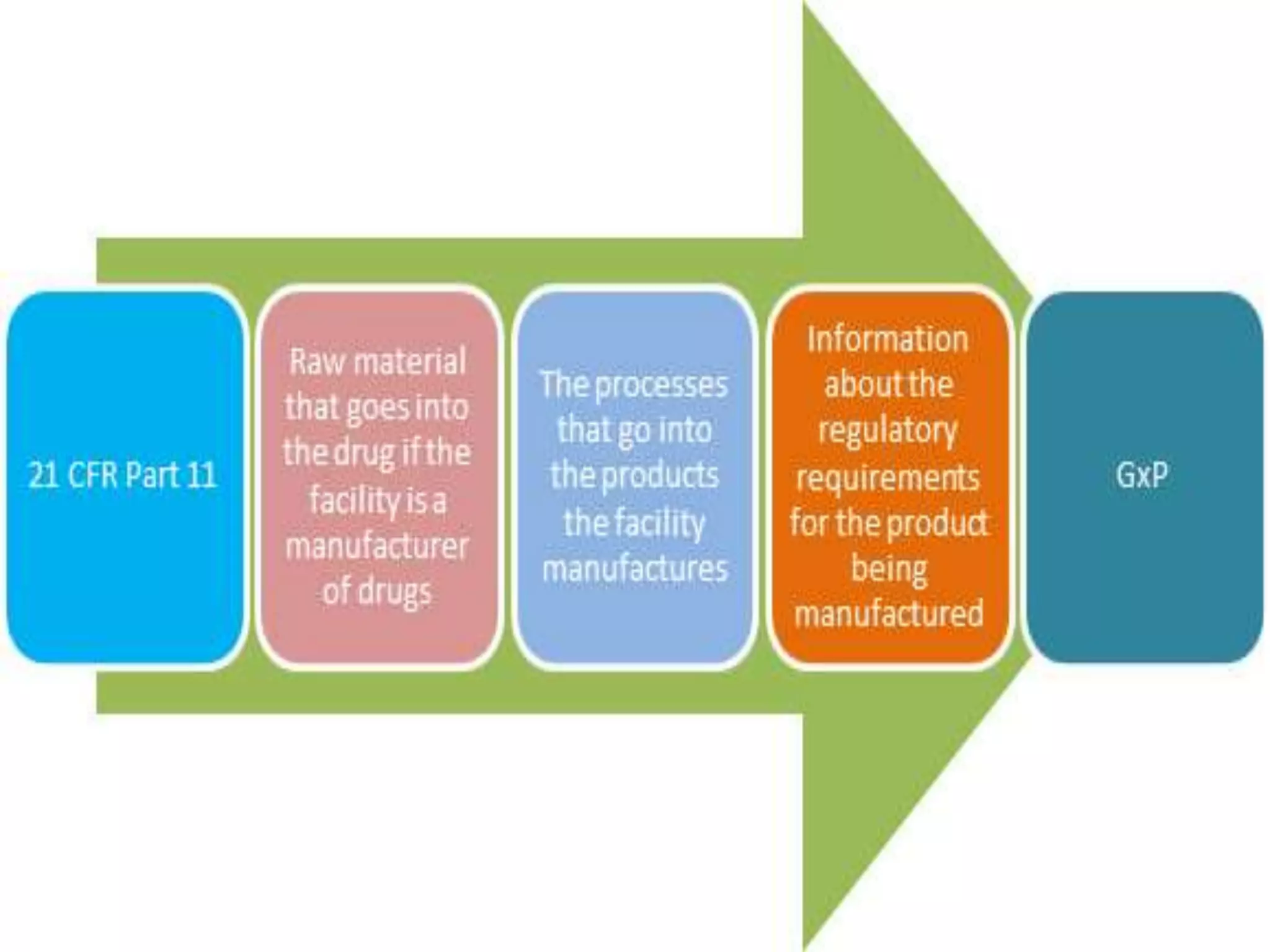
21 CFR PART11
CFR stands for Code of Federal Regulations and refers to a
document listing United States Federal Regulations.
The number "21" is short for "Title 21, Chapter I," and the
number "11," for "Part 11“.
Title 21 concerns the area of Food and Drugs, Chapter I is the
section related to FDA, and Part 11 is the sub-section of this
chapter, which focuses on a specific area (i.e., Electronic
Records; Electronic Signatures)
11
12.
IMPORTANCE OF 21CFR PART11
Guidelines on how to manage electronic records and
electronic signatures in order to maintain accuracy and
security.
Help FDA-regulated companies obtain the benefits of
electronic data management.
Prevent fraud while permitting the widest possible use of
electronic technology.
Guidelines that establish which electronic records and
signatures can be considered equivalent to paper records and
handwritten signatures.
12
13.
Part 11requires:
(1) Controlled access;
(2) Computer generated audit trails;
(3) Electronic digital signatures.
13
ELECTRONIC RECORDS:
Any combinationof text, graphics, data, audio, pictorial, or
other information representation in digital form that is
created, modified, maintained, archived, retrieved, or
distributed by a computer system.
ELECRONIC SIGNATURE:
A computer data compilation of any symbol or series of
symbols executed, adopted, or authorized by an individual to
be the legally binding equivalent of the individual's
handwritten signature.
15
16.
ELECTRONIC RECORDS
Secureprocess values and audit trails .
Protection of data through binary, compressed and check-
summed records .
Accurate time stamps are ensured using automatic Time
Synchronization to a known clock source .
Provision for electronically copying data for archive .
Export facility providing viewing of secure records in human
readable form.
16
17.
ELECTRONIC SIGNATURE
Alluser actions can be configured to require signing or require
signing and authorization .
User specific access according to authority level .
controls unique user signature, password expiry, minimum
password length, automatic log-off, automatic disabling and
notification of failed login attempts .
Ensuring unique users by retiring and not deleting accounts
17
18.
ADVANTAGES
Electronic Batchrecords can eliminate mountains of paper
work, speed processing and allow for statistical and trend
analyses.
NDA’s and other submissions can be submitted electronically
in place of paper submission.
Increases the speed of information exchange.
Cost savings from reduced need for storage space.
Manufacturing process streamlining.
Job creation in industries involved in electronic record and
electronic signature technologies.
18
19.
APPLICATIONS
Section 11.10describes controls for closed systems.
These controls designed to ensure the integrity of system
operations and information stored in the system.
Section 11.30 describes controls for open system .
Develop procedures and controls that ensure authenticity,
integrity, and confidentiality of electronic records and comply
with all other parts of Section 11.10
Must use additional measures to ensure authenticity,
integrity, and confidentiality. 19
20.
20
Section 11.70:
Signature /record linking
Electronic signature and handwritten signatures must be
linked to ensure signatures cannot be excised, copied,
transferred or falsified.
Section 11.50:
It requires signature manifestations to contain information
associated with the signing of electronic record.
Signed electronic records must include :
printed name of the signer
date and time of signature
the purpose of the signature (e.g. review, approval etc.)
Each of these must be readable by display or printout.
21.
21
Section 11.100:
Mustbe unique to an individual and not reassigned
Identity of individual must be verified by organization
Must certify electronic signature system to the agency prior to
or at the time of use of the system
Certification must be submitted in paper form and, upon
agency request, provide certification that signature is legally
binding
22.
22
Section 11.200:
ElectronicSignature Components and Controls.
Non biometric signatures must:
Contain at least two different identification components
Be used only by the owner
Ensure use by other individuals is precluded and does not
occur without collaboration by at least two other
individuals
Biometric signatures must ensure use by the owner.
23.
23
Section 11.300:
Controlsfor Identification Codes/Passwords.
Persons using electronic signatures must use controls to
ensure security and integrity should include:
Assuring that no two individuals have the same
combination of identification code and password
Periodic check, recall of code and password
Loss management and replacement procedures.
Unauthorized use safeguards
Report attempts in urgent & immediate manner
Security unit
Management, as appropriate
24.
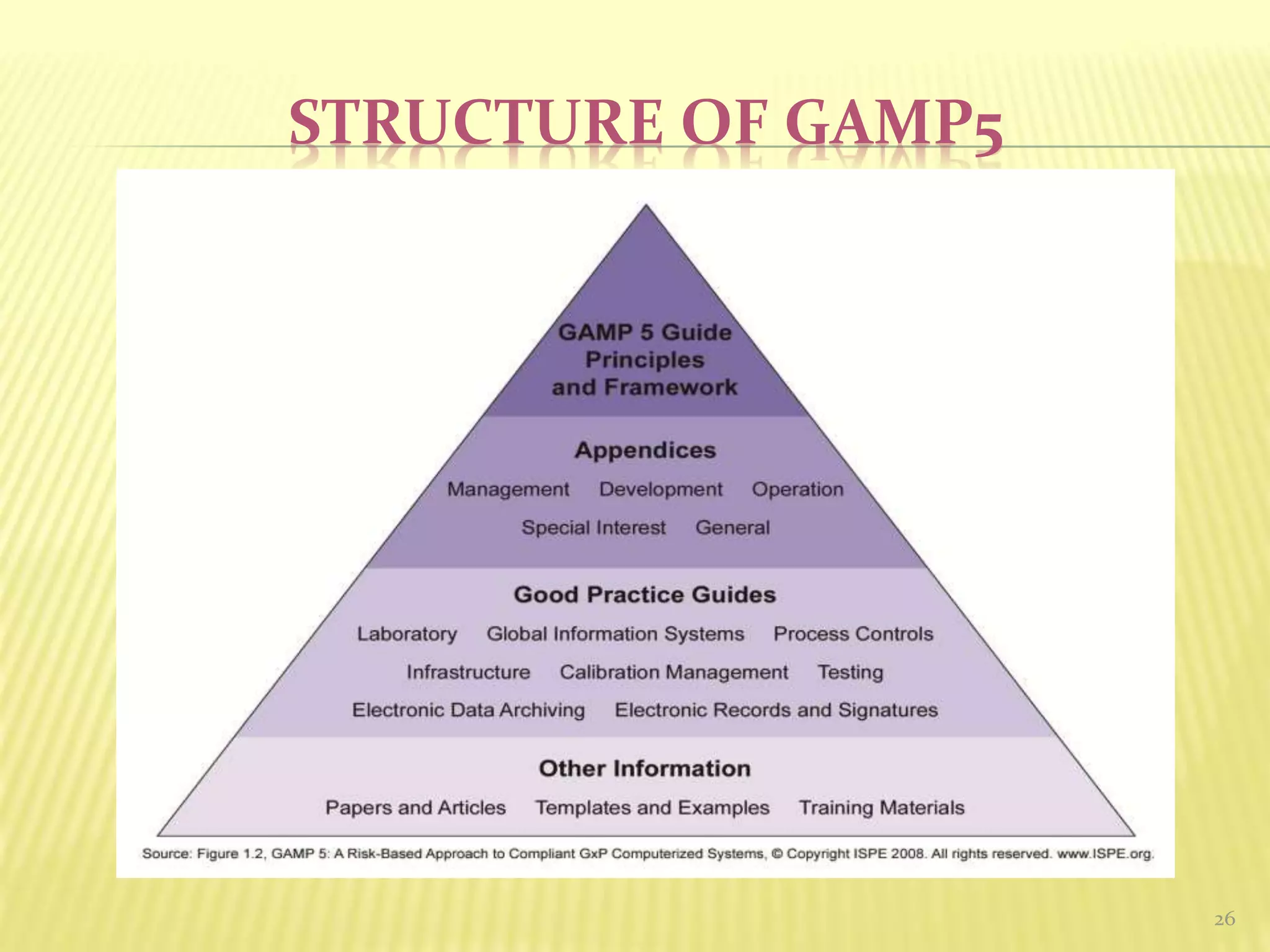
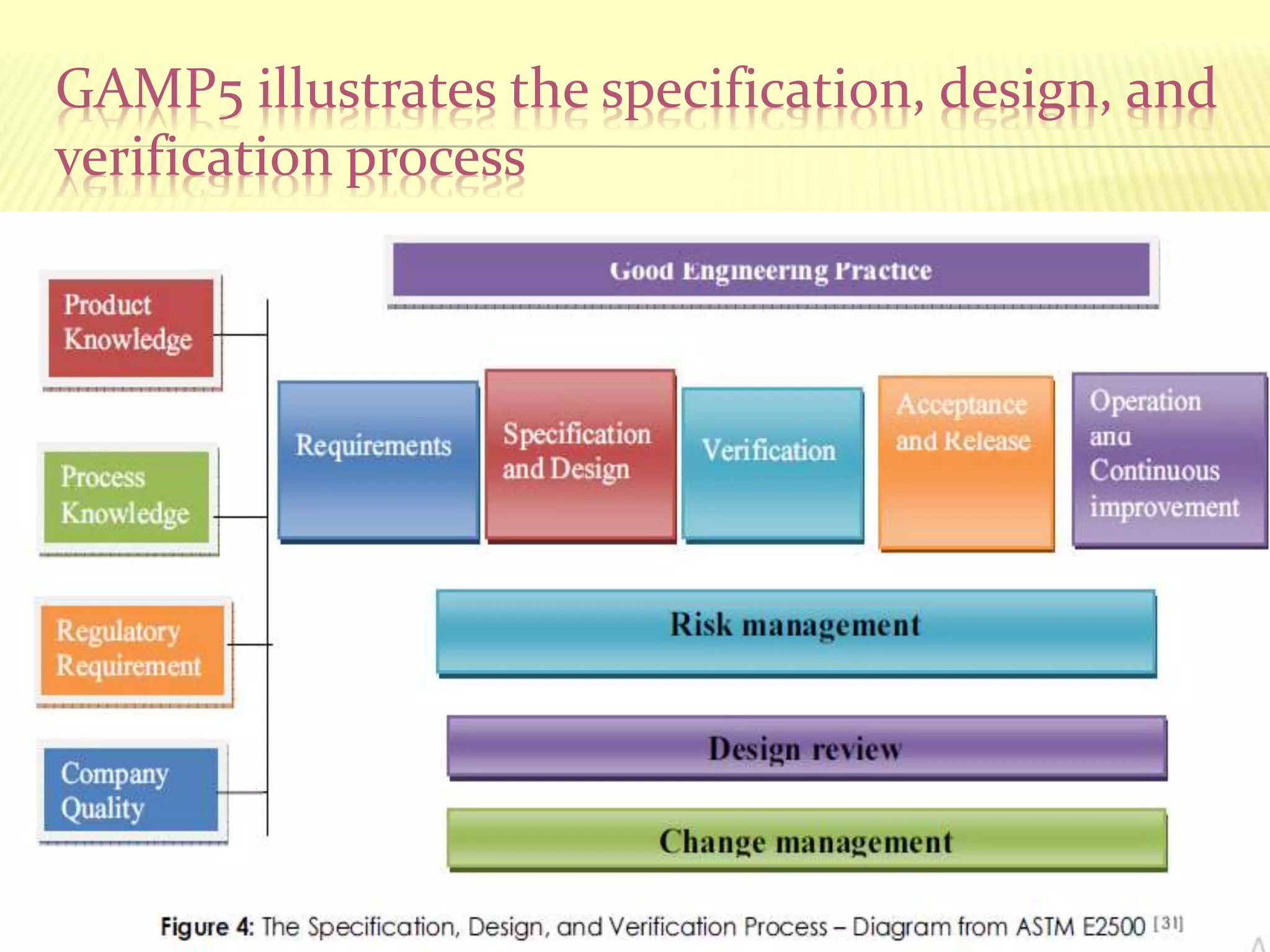
GAMP 5
GAMP5guidance aims to achieve computerized systems that
are fit for intended use and meet current regulatory
requirements, by building upon existing industry good
practice in an efficient and effective manner.
24
25.
The purpose ofthe guidelines is to:
Provide a cost effective framework of good practice to ensure
that computerized systems are fit for use and compliant with
regulation.
Key concepts to GAMP 5:
Product and Process Understanding.
Lifecycle approach within QMS.
Scalable Lifecycle Activities.
Science Based Quality Risk Management.
Leveraging Supplier Involvement.
25
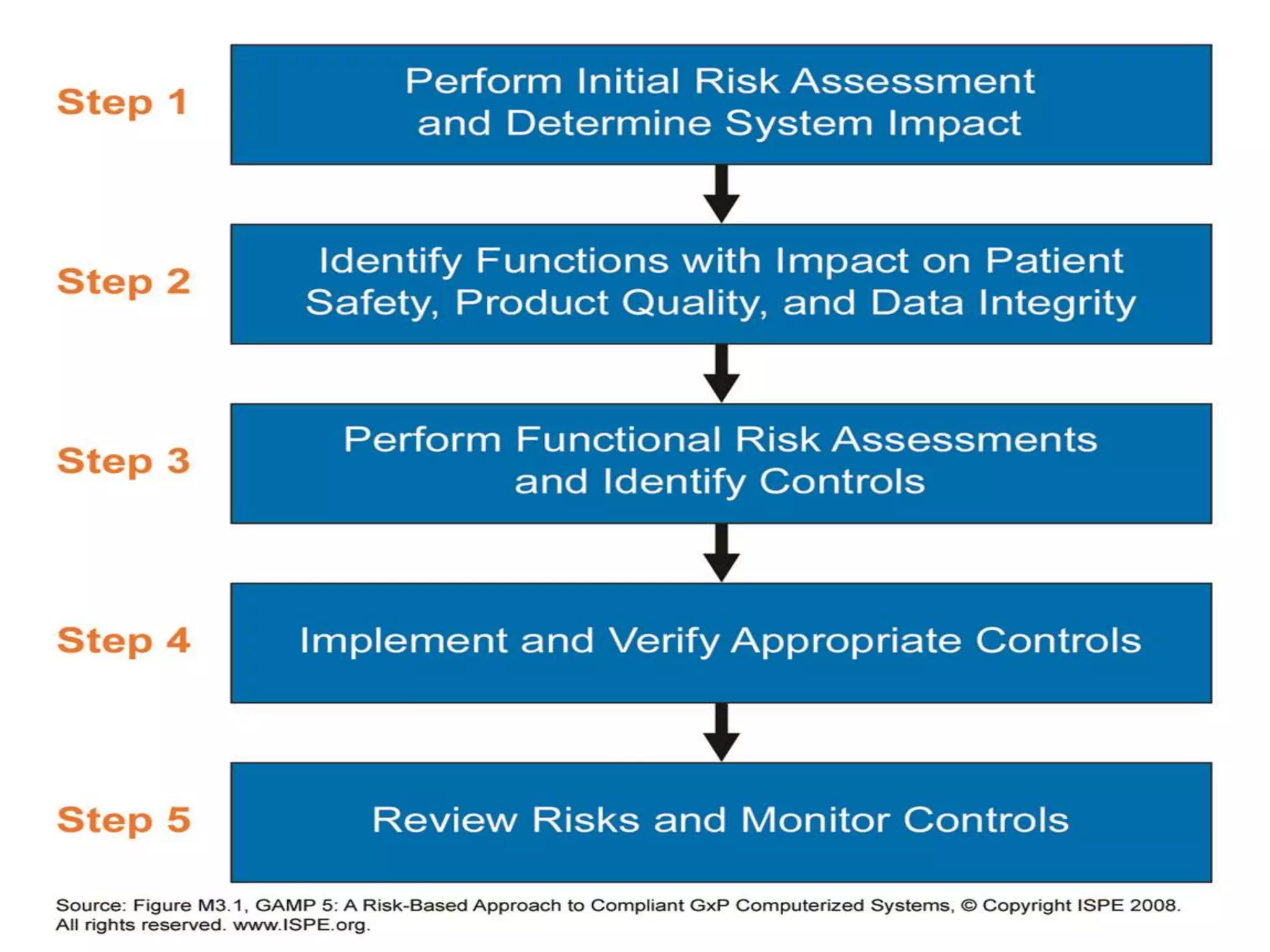
GAMP 5sets the main requirements for the use of
computerized systems in pharmaceutical applications:
Patient safety, product quality and data integrity.
Effective governance to achieve and maintain GxP
compliance.
Quality by design (QBD).
Continuous improvement with in QMS.
Critical quality attributes (CQA).
28
29.
Improving GXPcompliance efficiency.
Configurable systems and development models.
Use of existing documentation and knowledge.
Effective supplier relationships.
Scalable approach to GXP compliance.
Science based quality risk management system.
Life cycle approach within QMS.
29
APPLICATIONS
1. Monitoring manufacturing,production and storage
environments in the pharmaceutical industry.
2. Monitoring the autoclaving process in the pharmaceutical
industry.
3. Water purification in the pharmaceutical industry.
4. Freeze drying in the pharmaceutical industry.
31