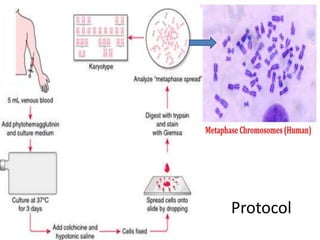
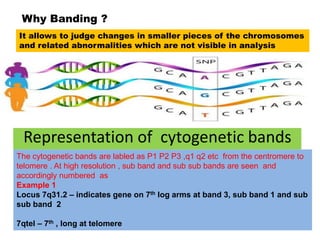
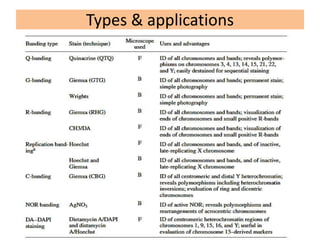
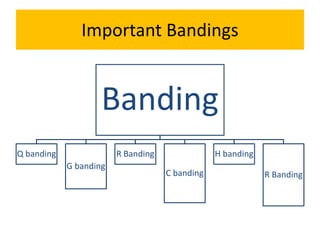
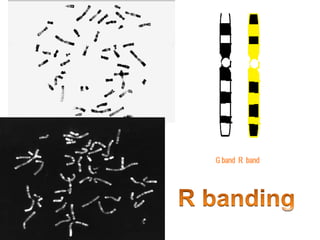
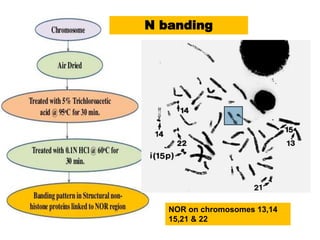
The document describes various chromosomal banding techniques used to analyze and classify chromosomes, focusing on karyotype preparation and banding methods such as G-banding, Q-banding, and R-banding. It details protocols for preparing chromosomal samples, staining procedures, and the significance of identifying chromosomal abnormalities through these techniques. Additionally, it addresses the implications of chromatin structure in cytogenetics and various staining methods to visualize chromosomal domains.