Download as PDF, PPTX



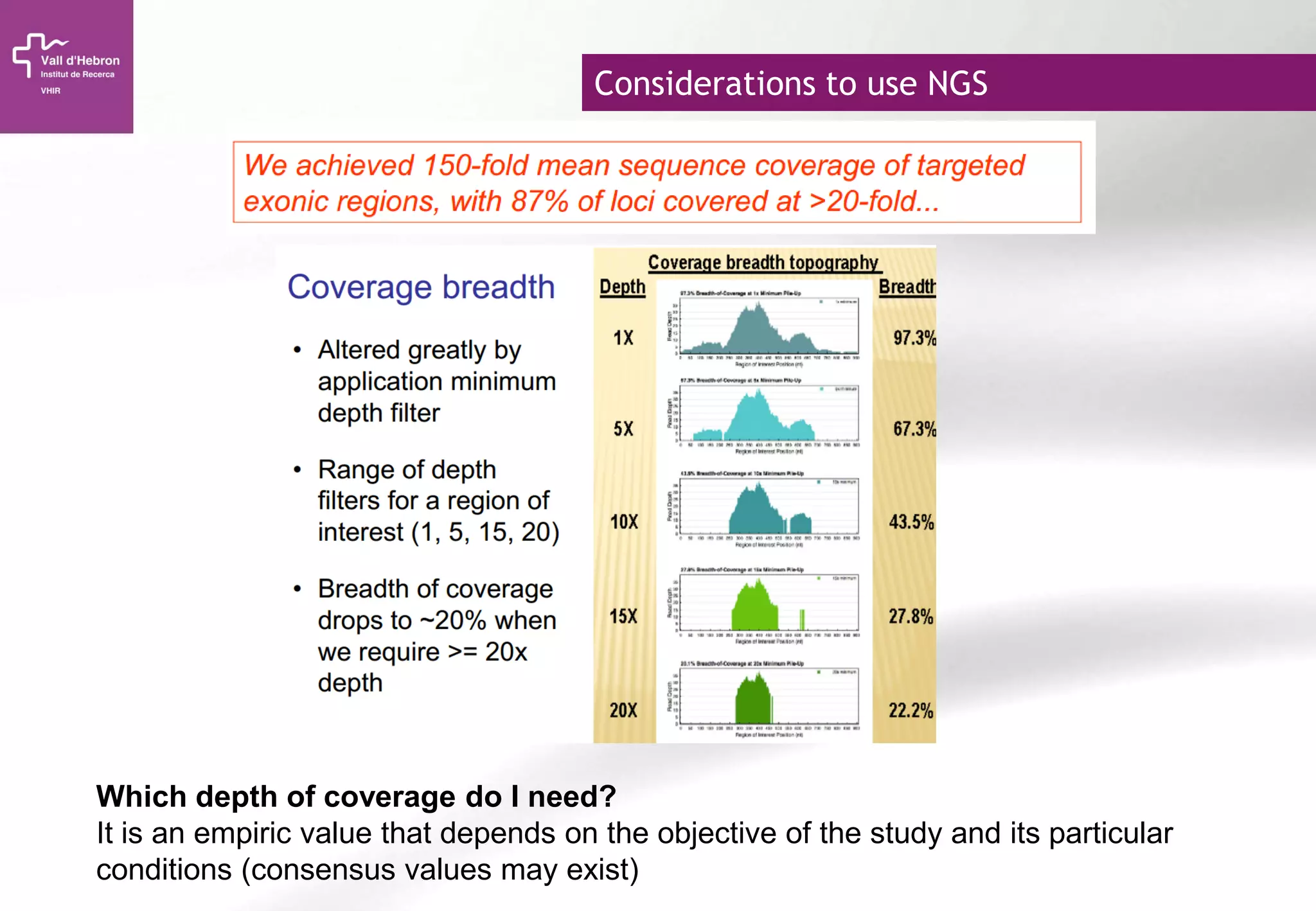


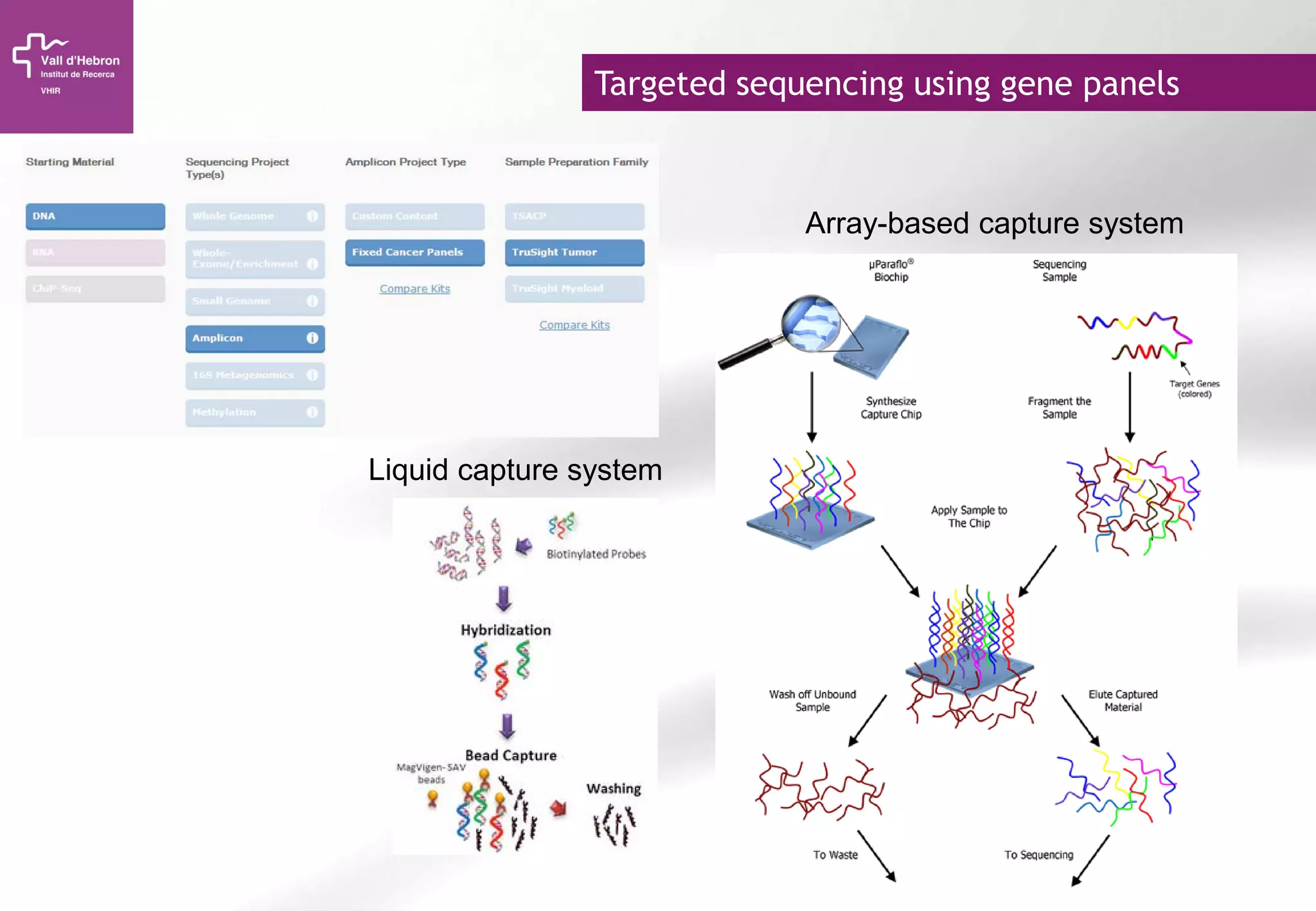


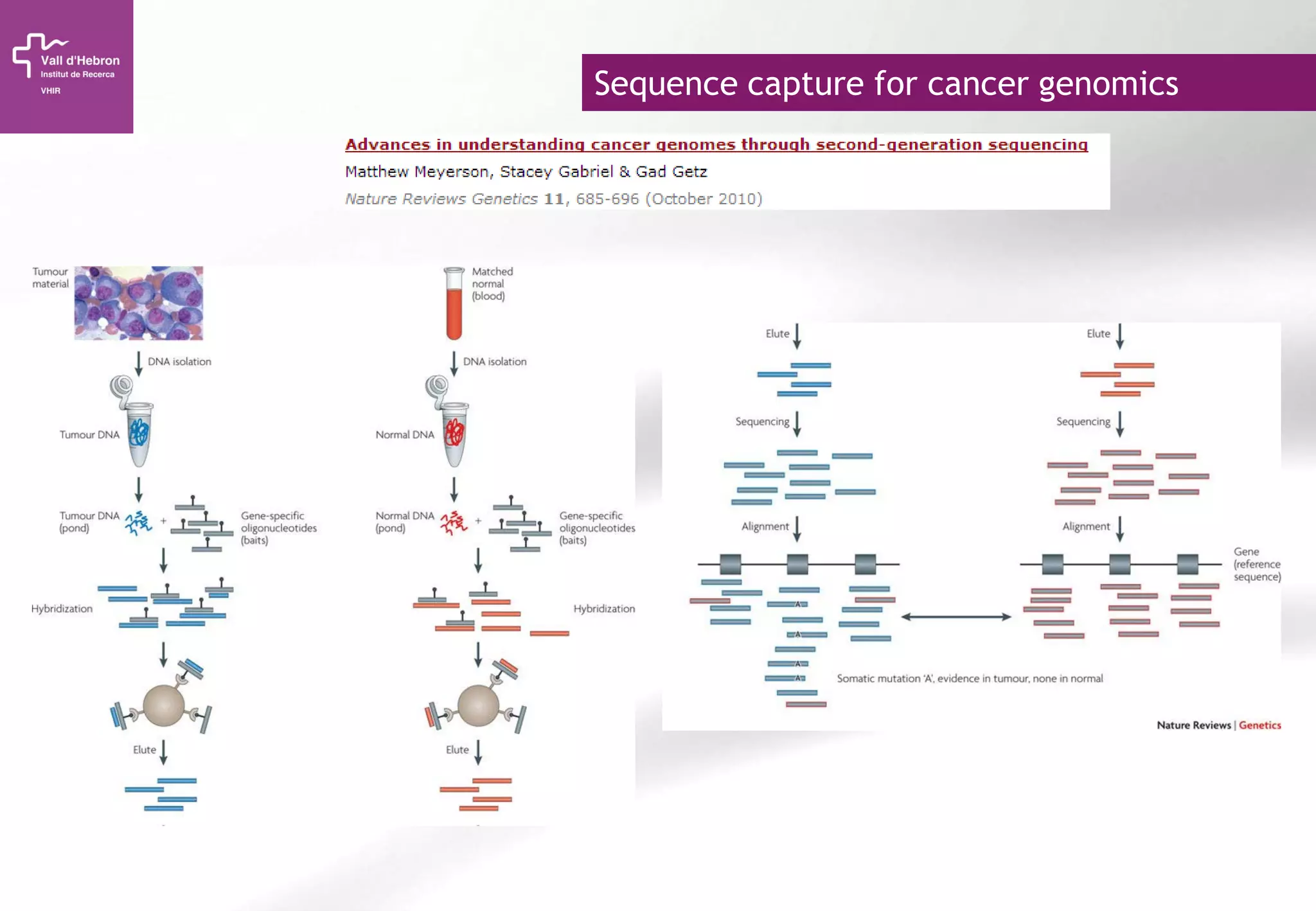

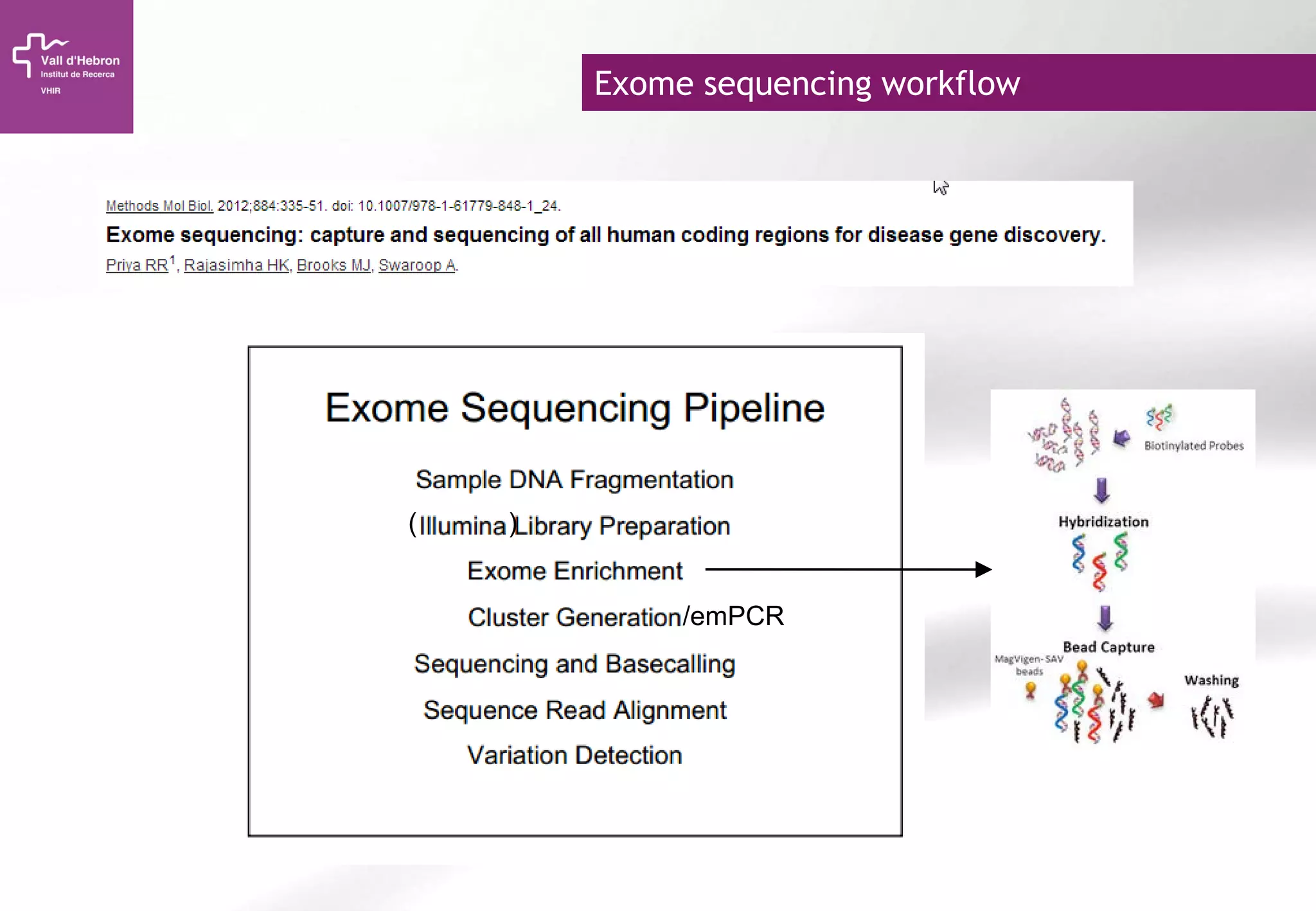

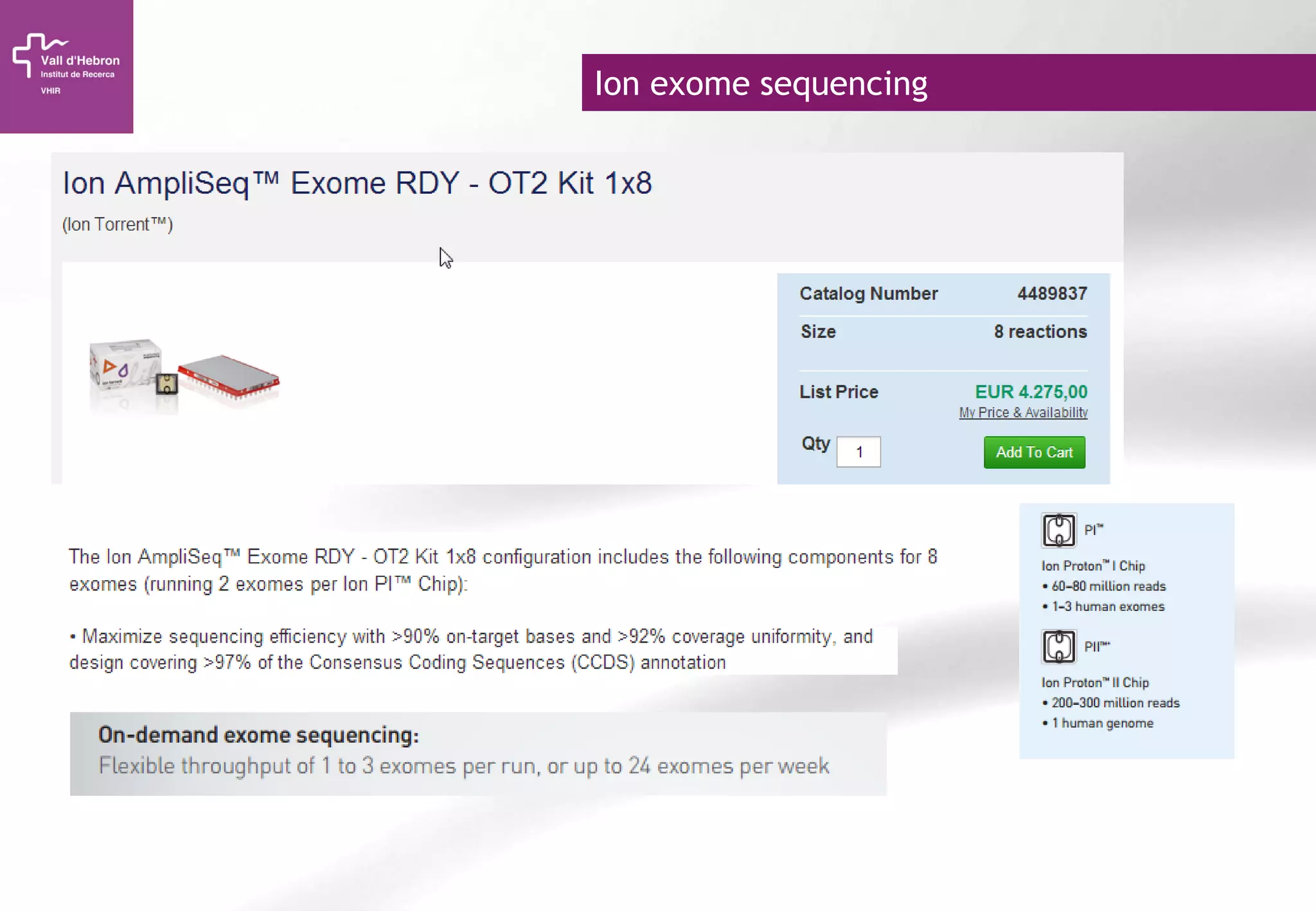

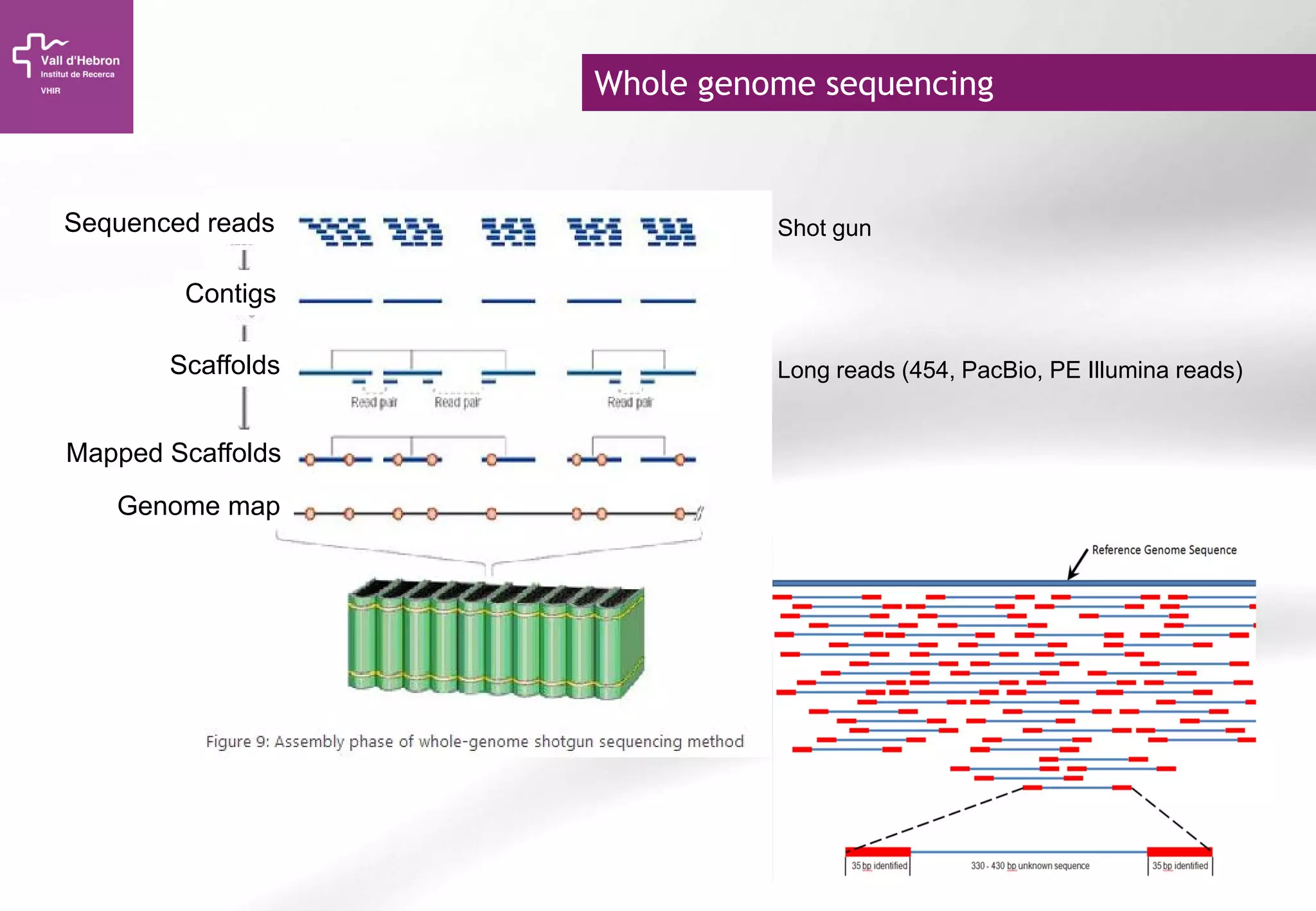
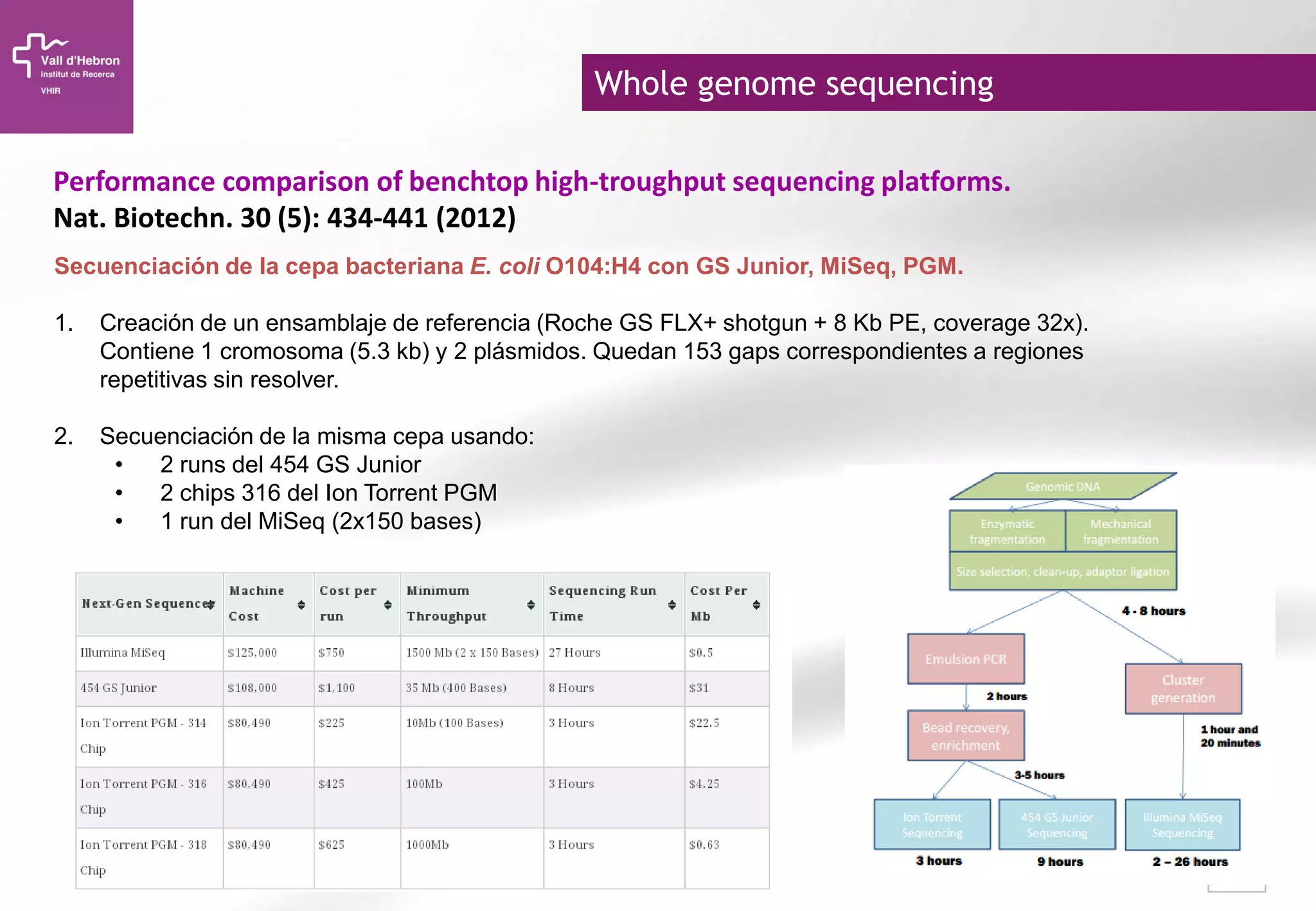



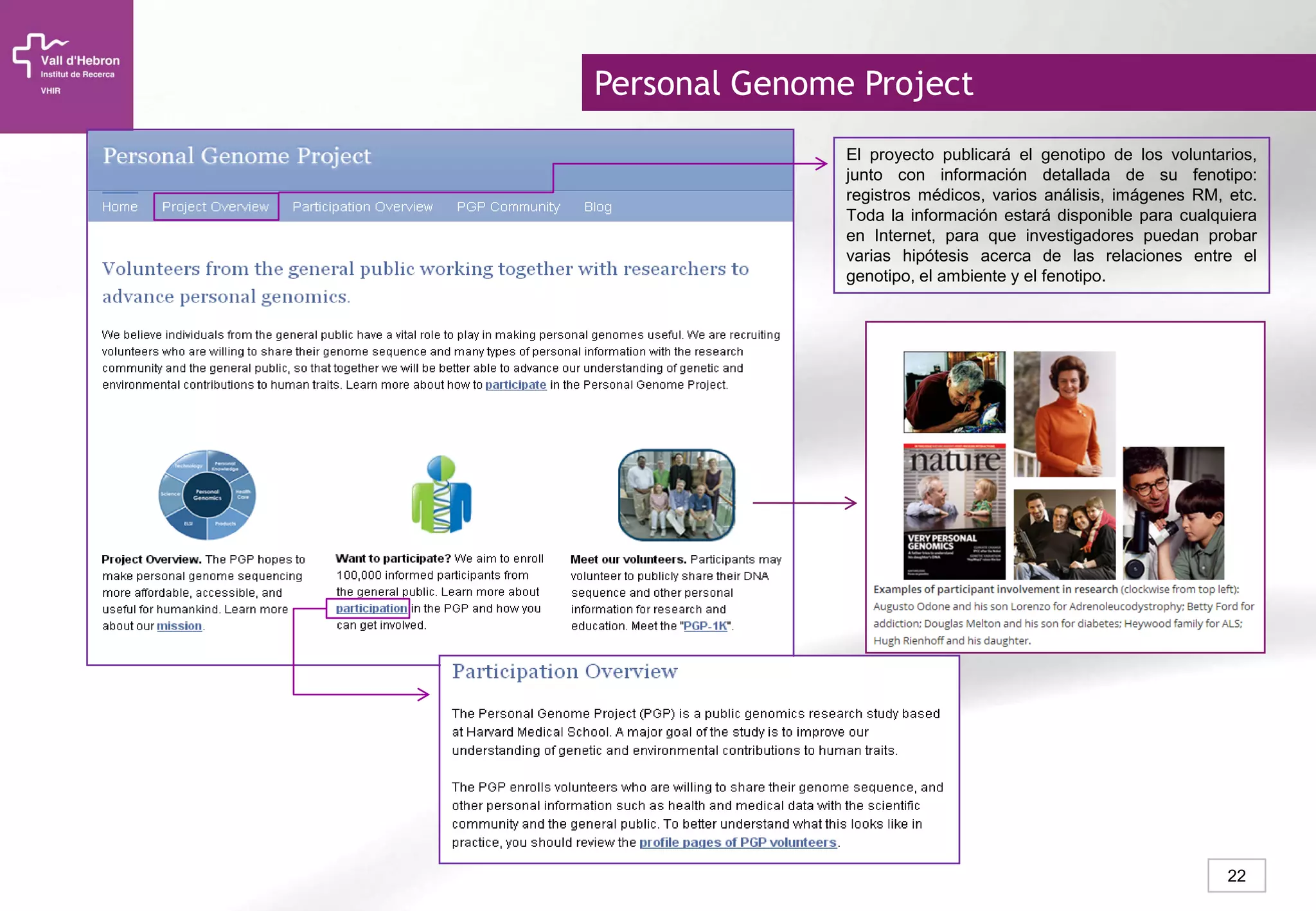

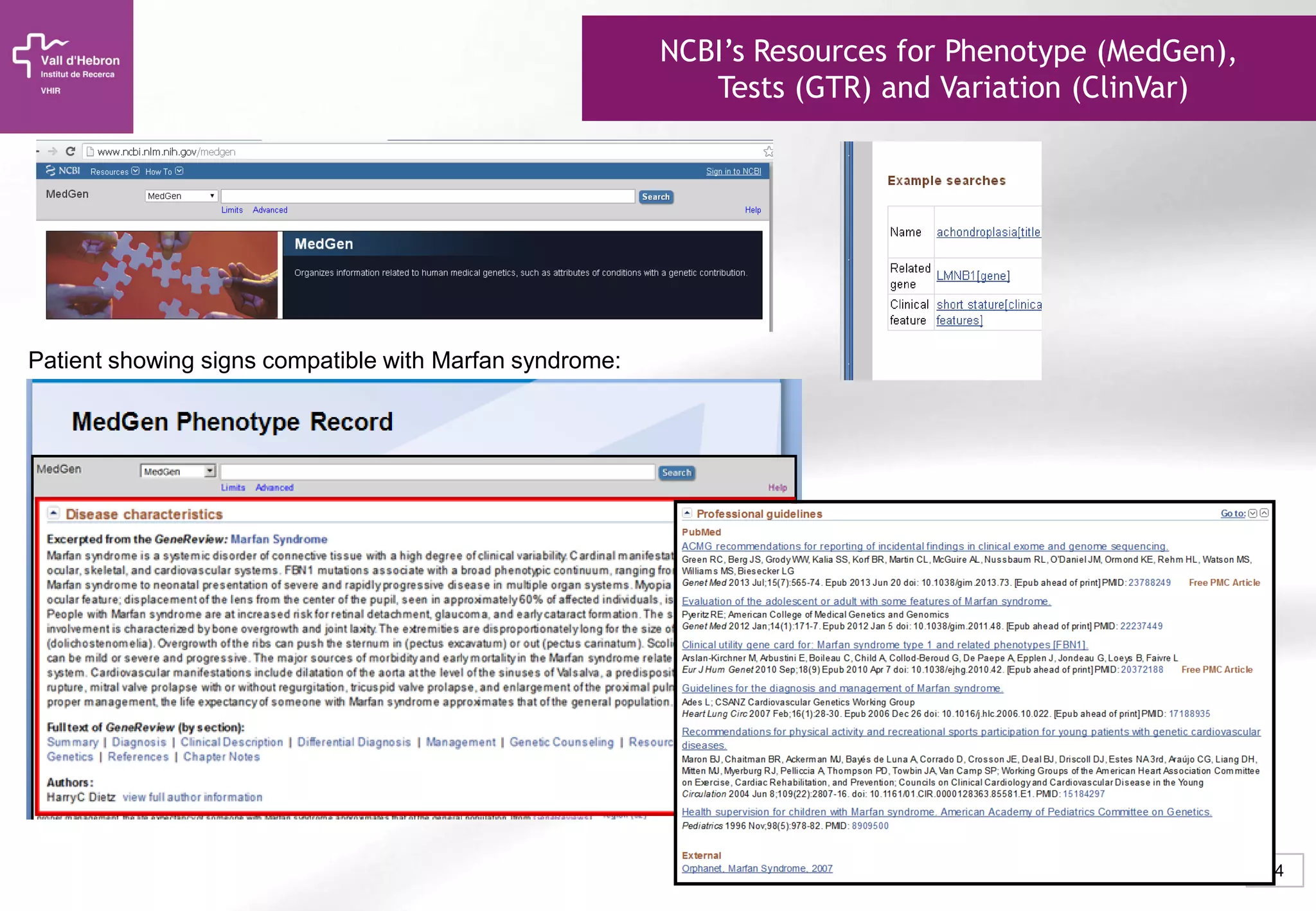
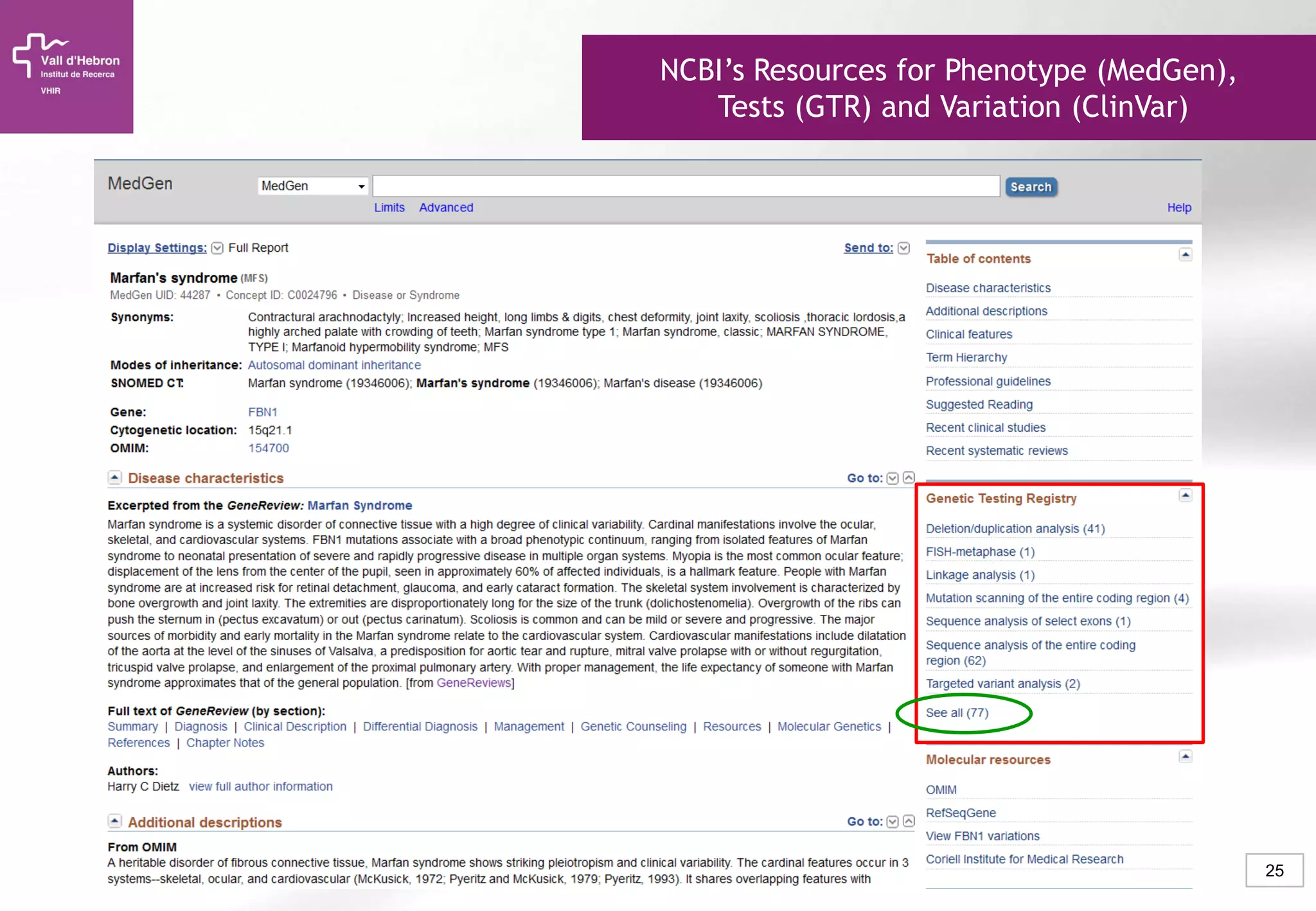
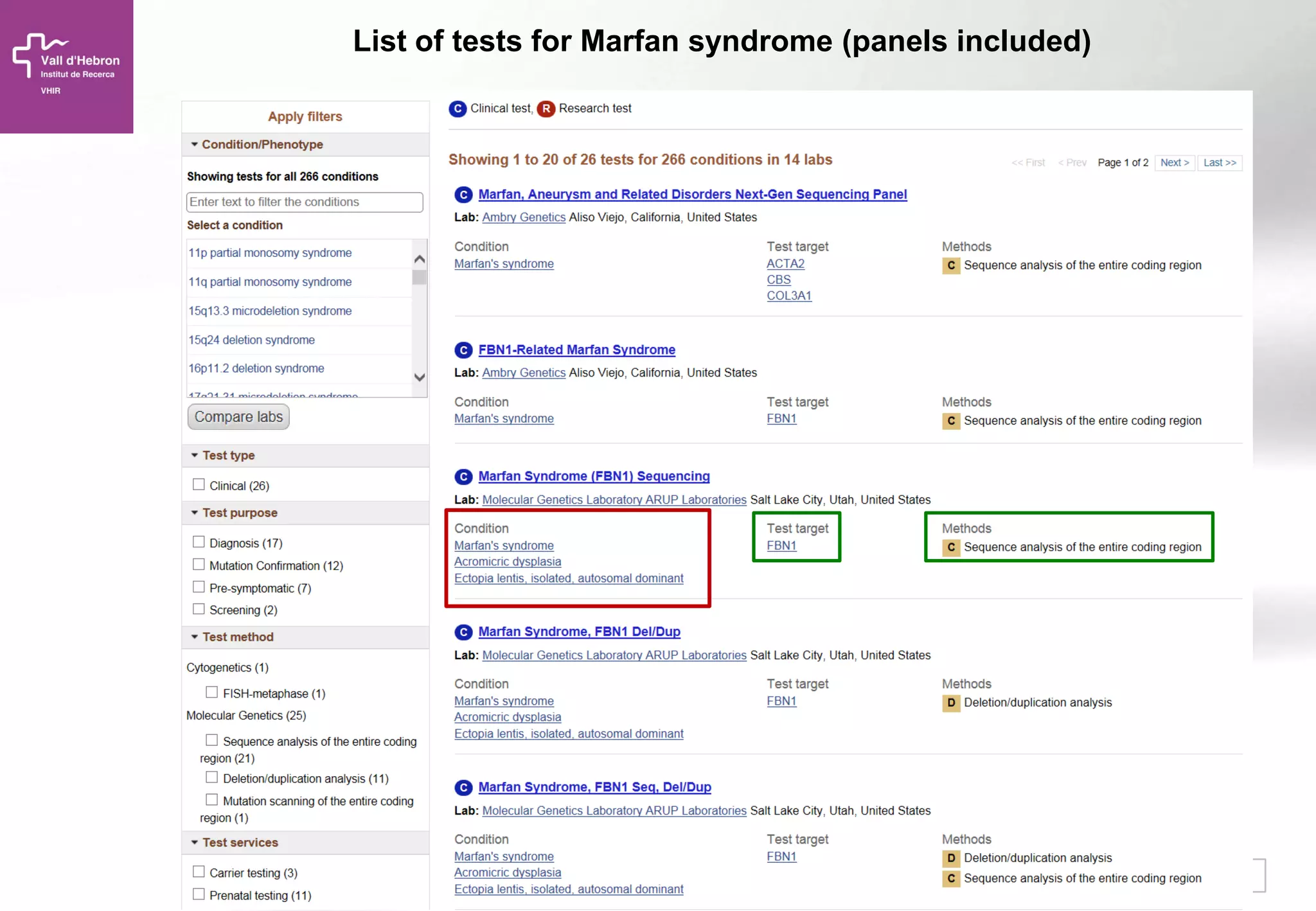
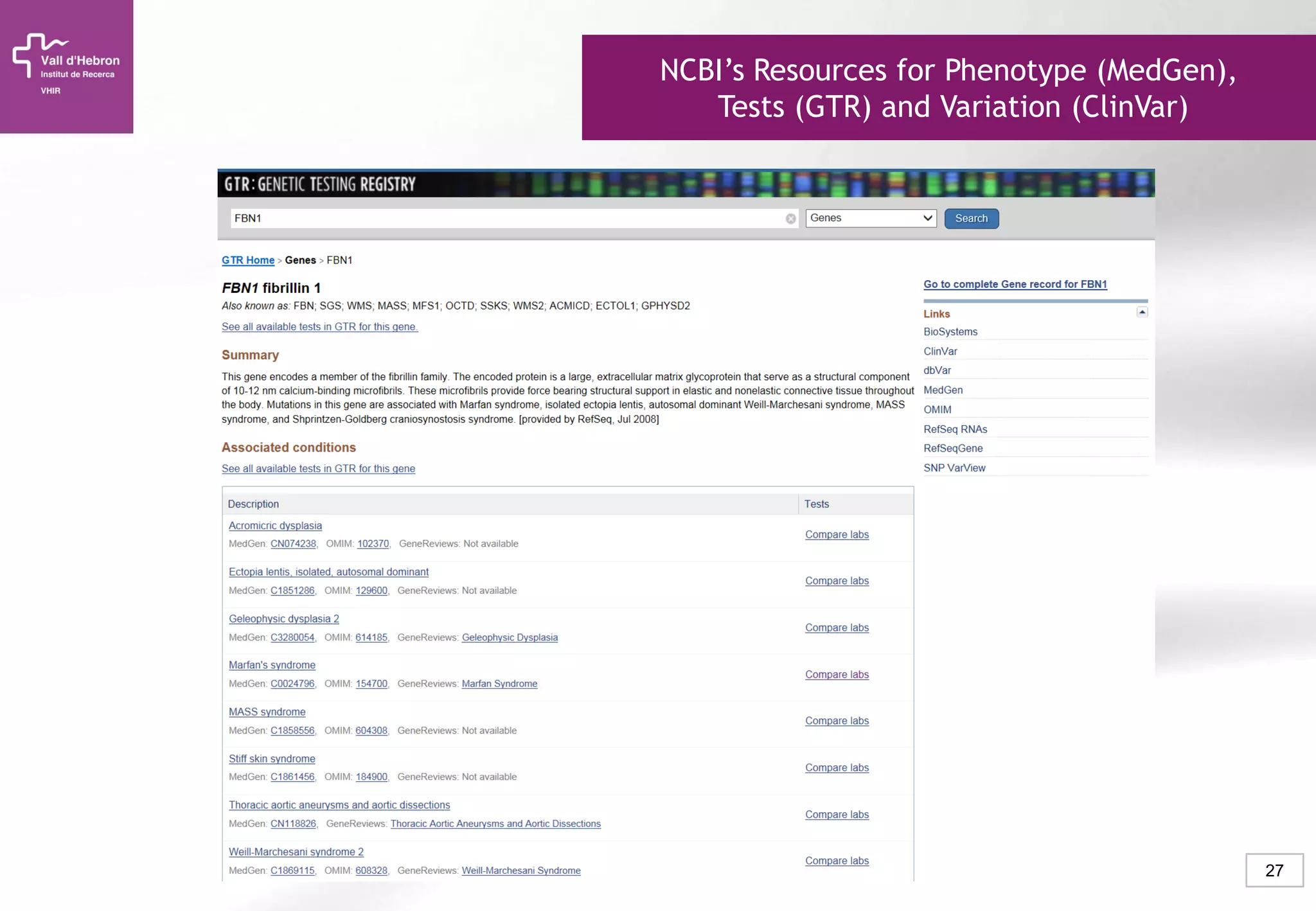
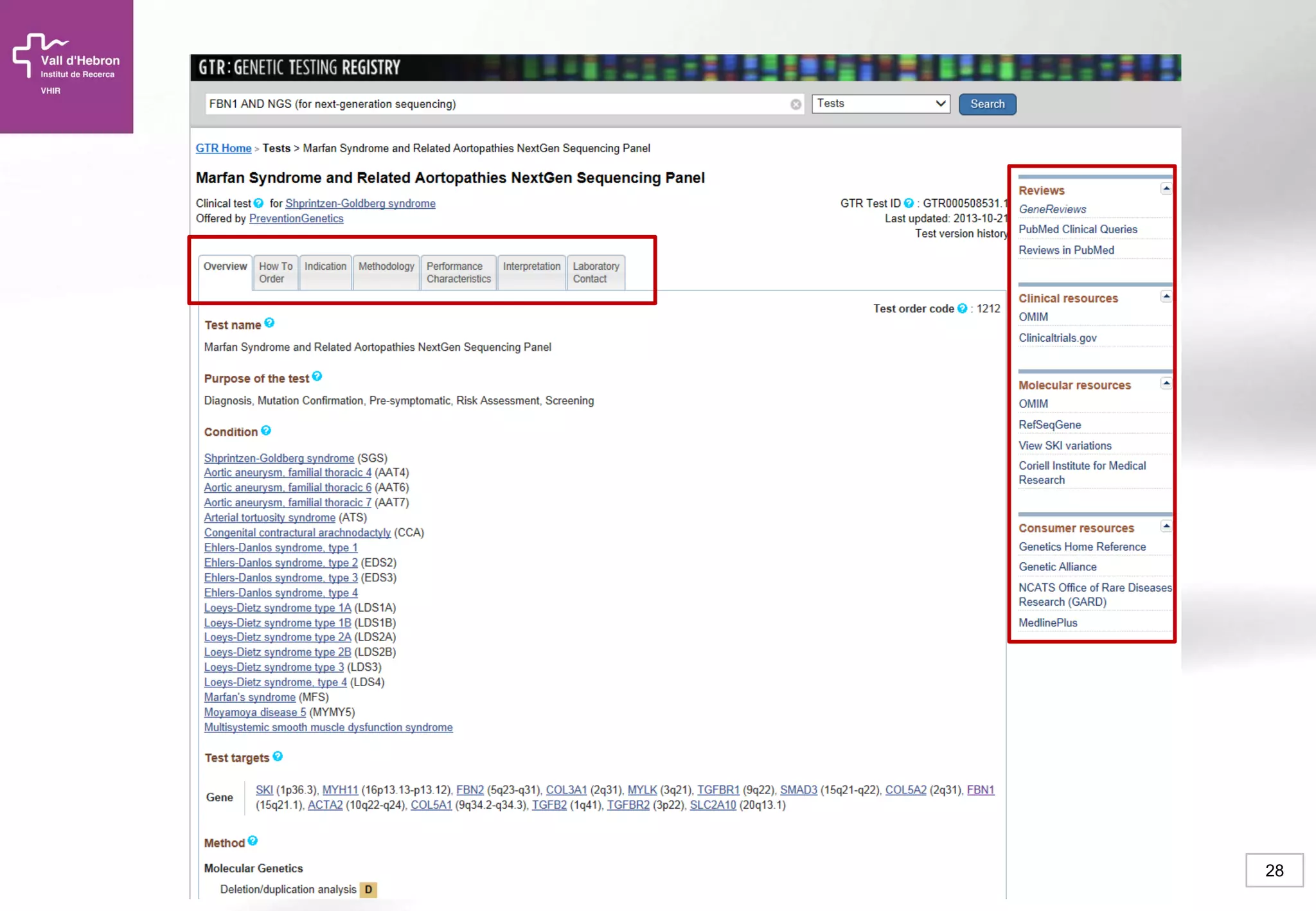
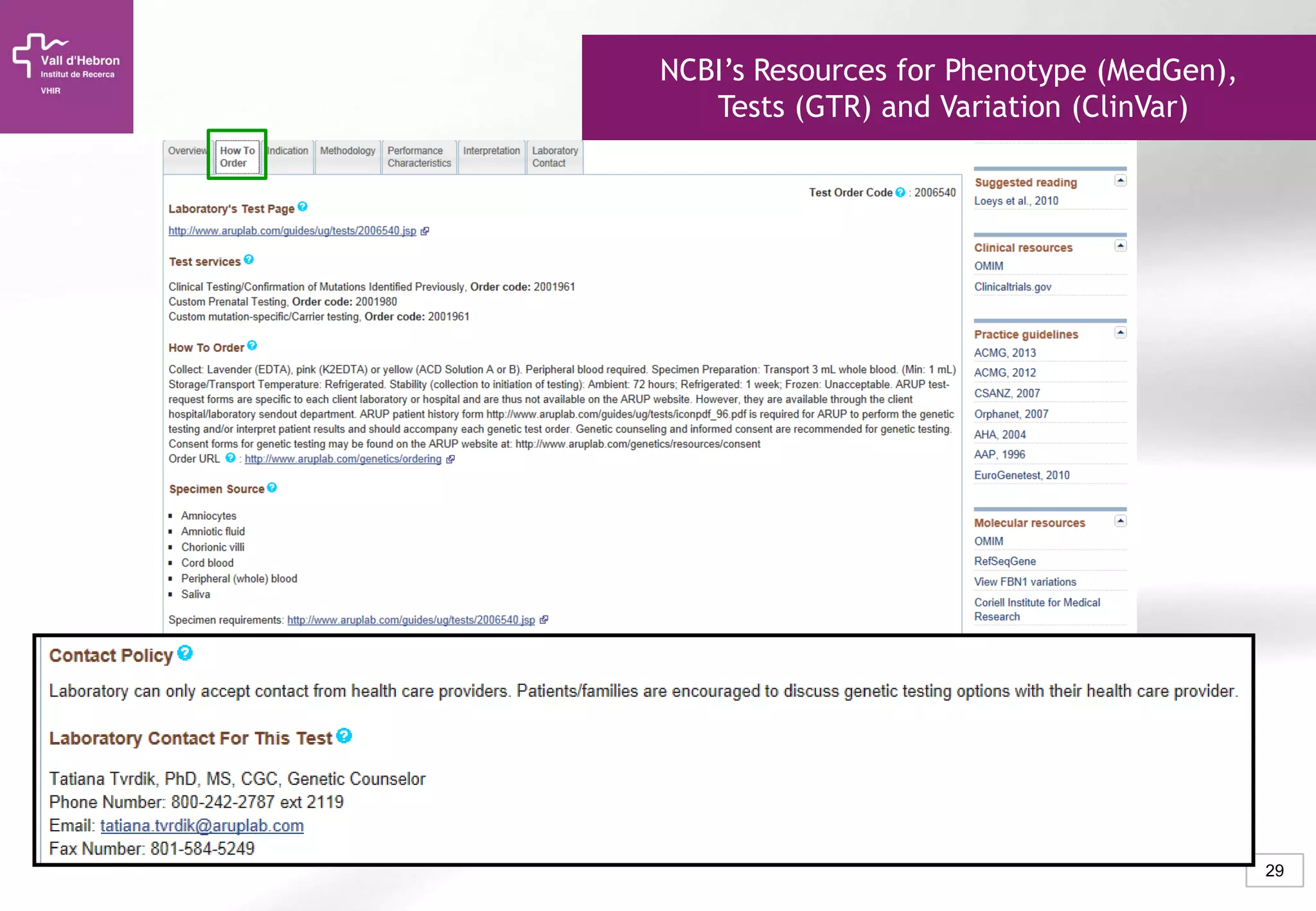
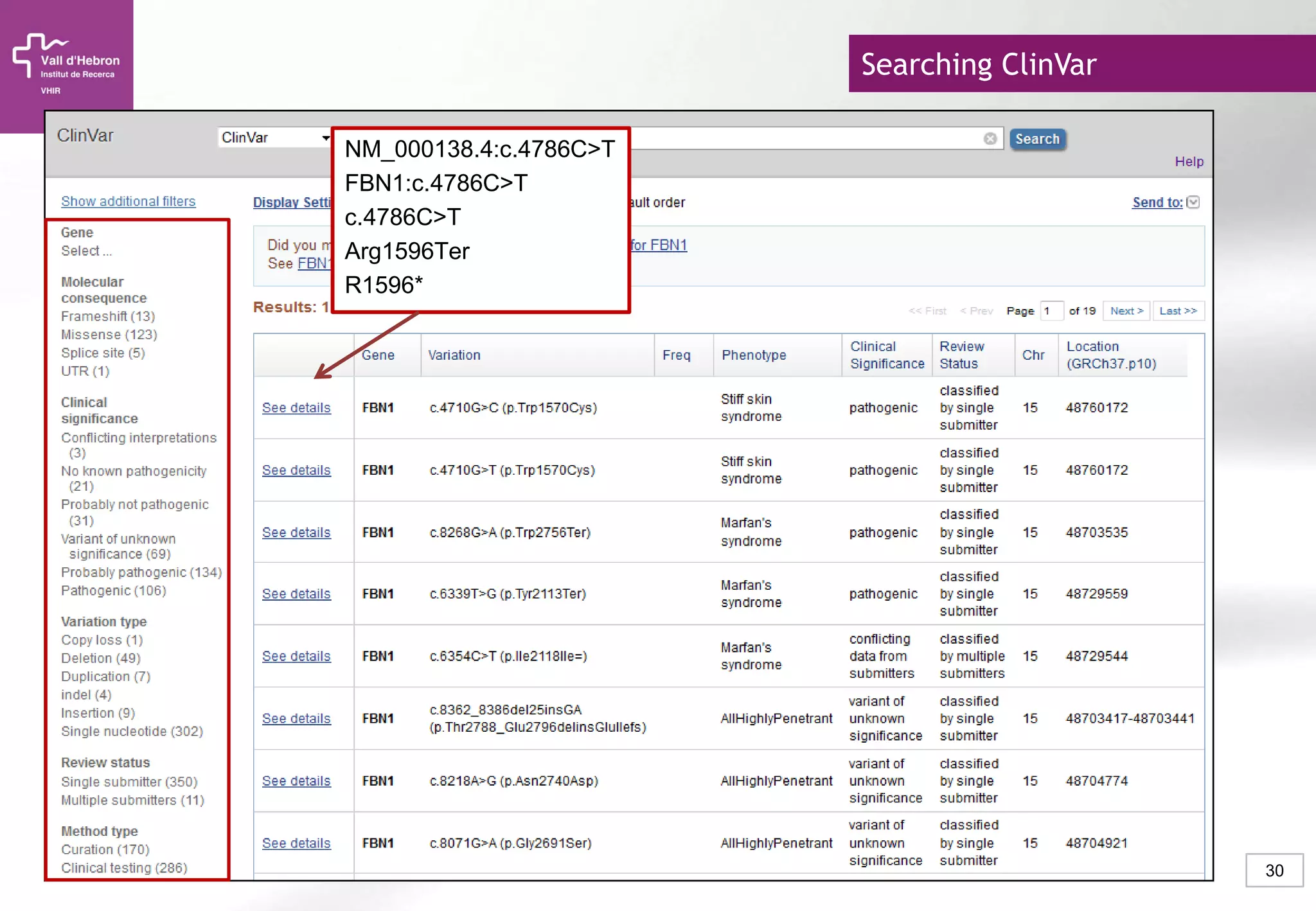
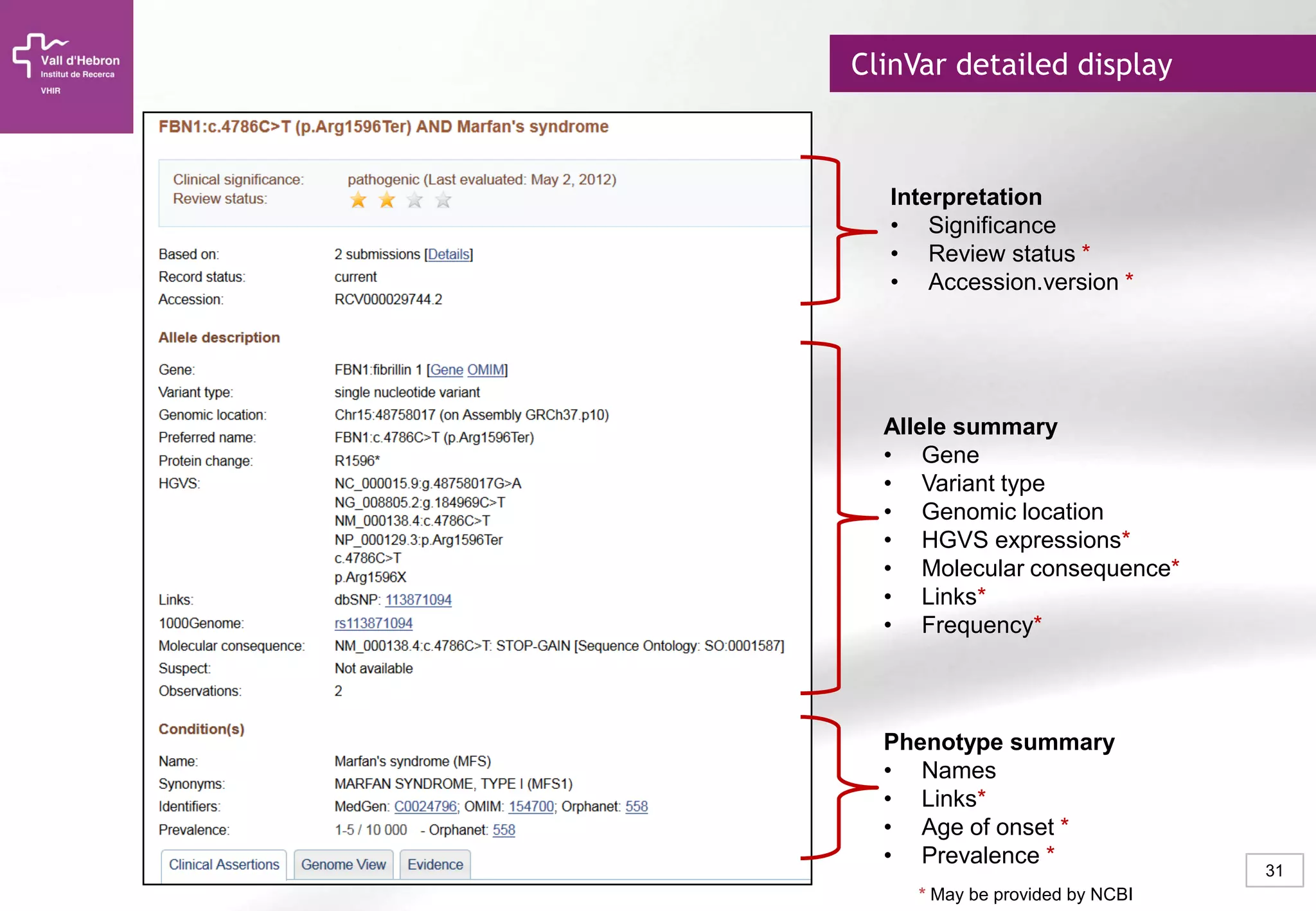
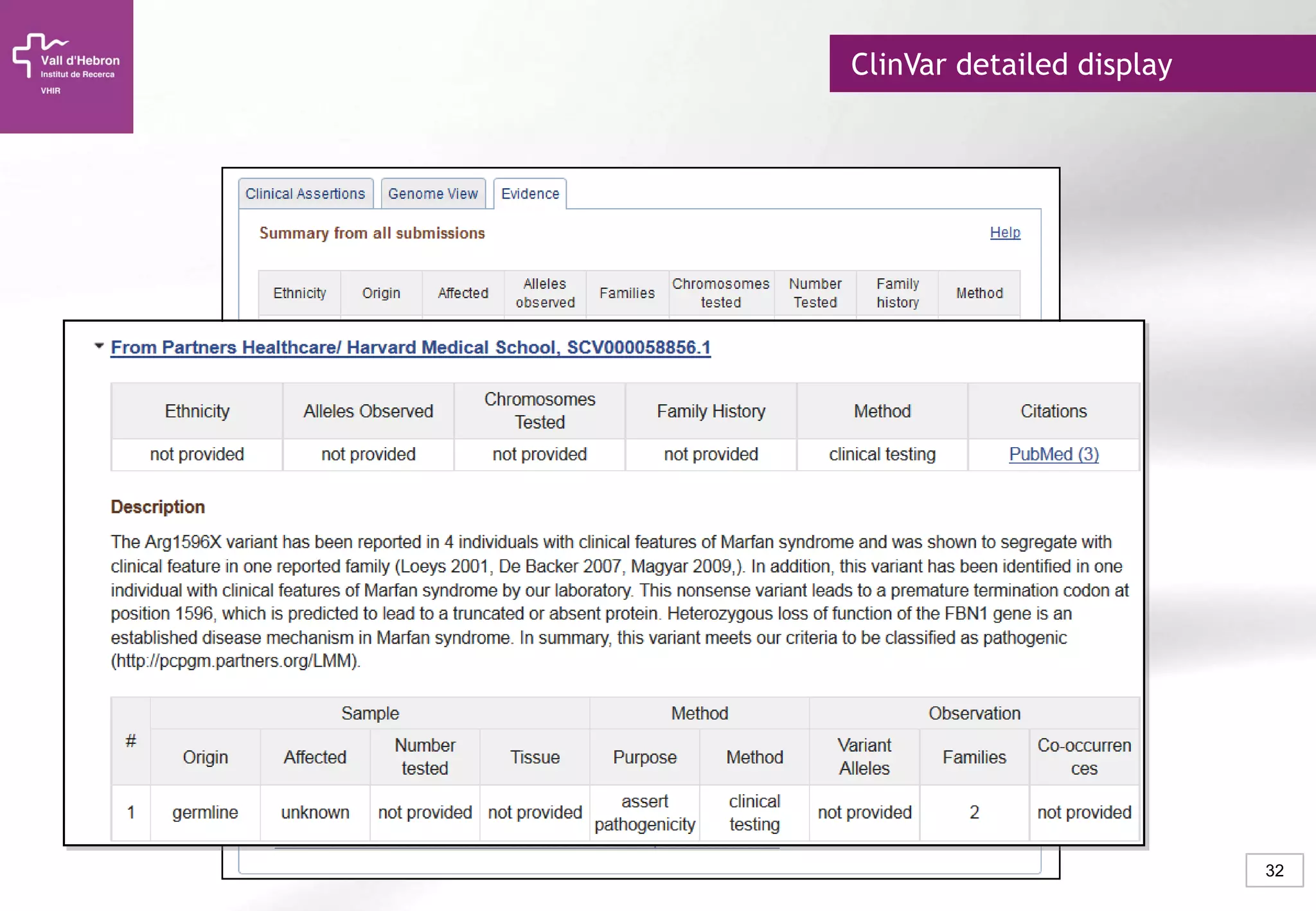

The document provides an overview of next-generation sequencing (NGS) technologies, their applications, and considerations for their use in biomedical research. It outlines various sequencing methods such as amplicon, targeted, exome, and whole genome sequencing, highlighting their unique advantages and challenges. The document also discusses the importance of NGS in identifying rare genetic variants and the impact of genetic diversity on health outcomes.











































































































