Downloaded 88 times



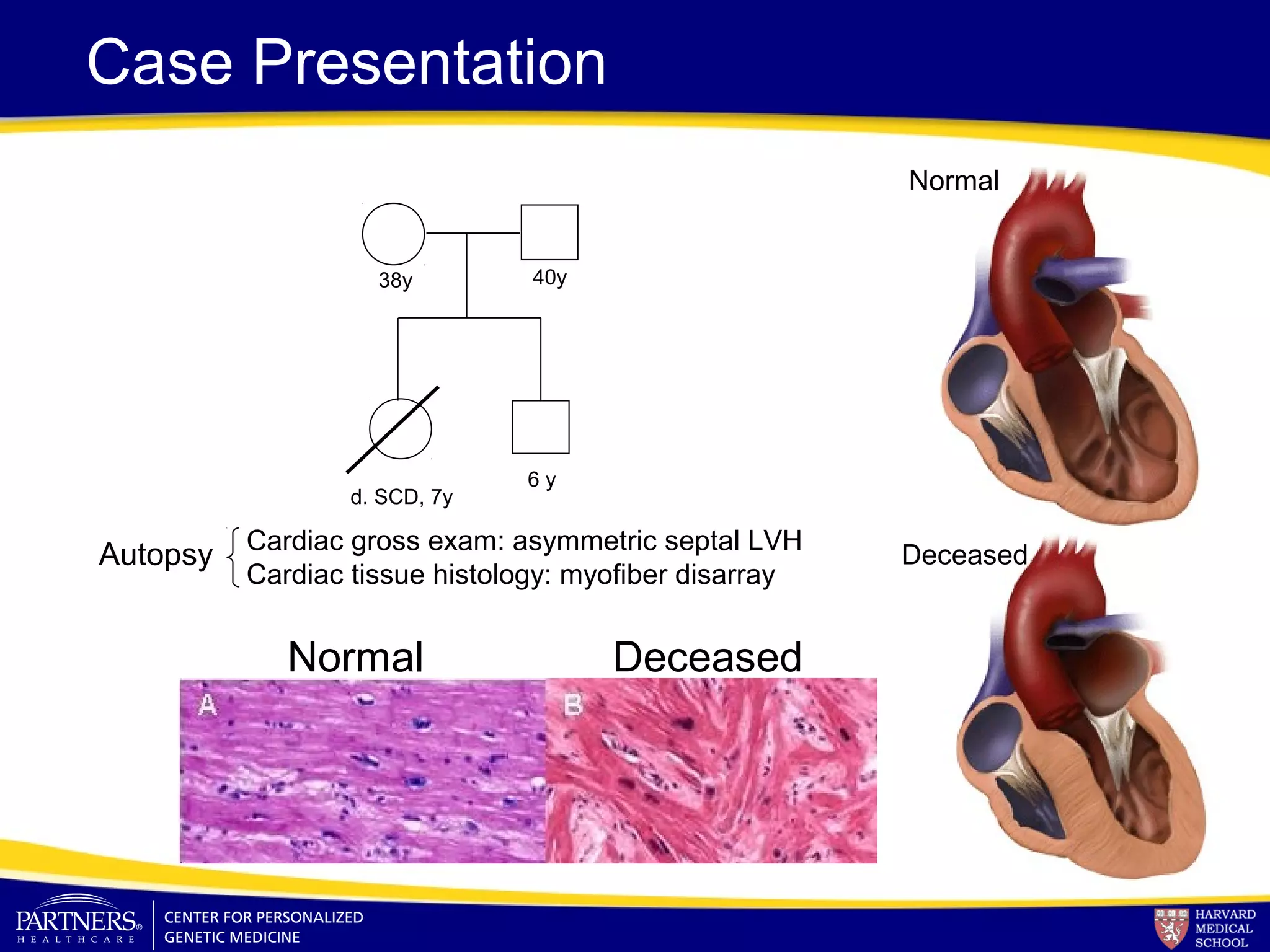
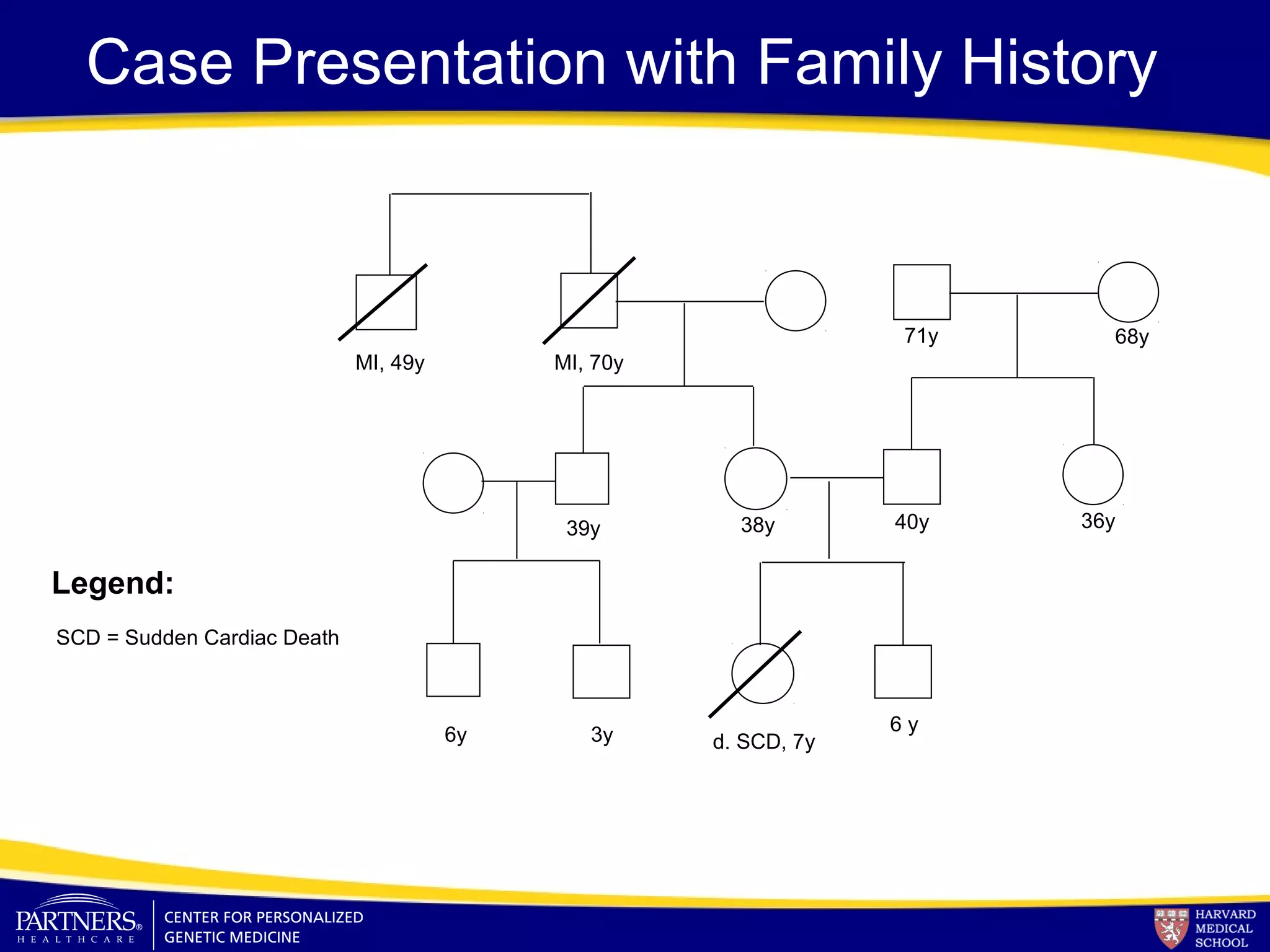
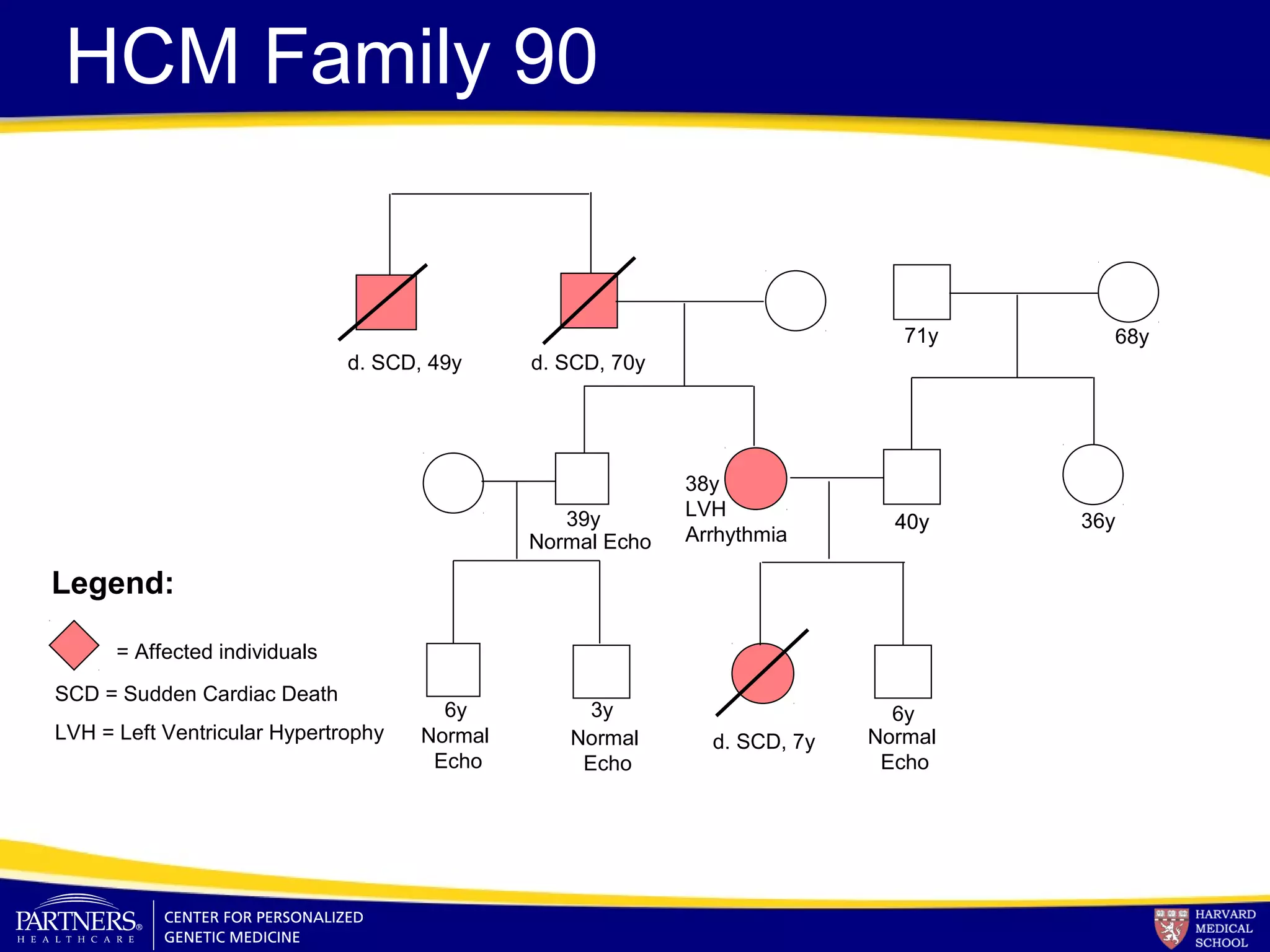
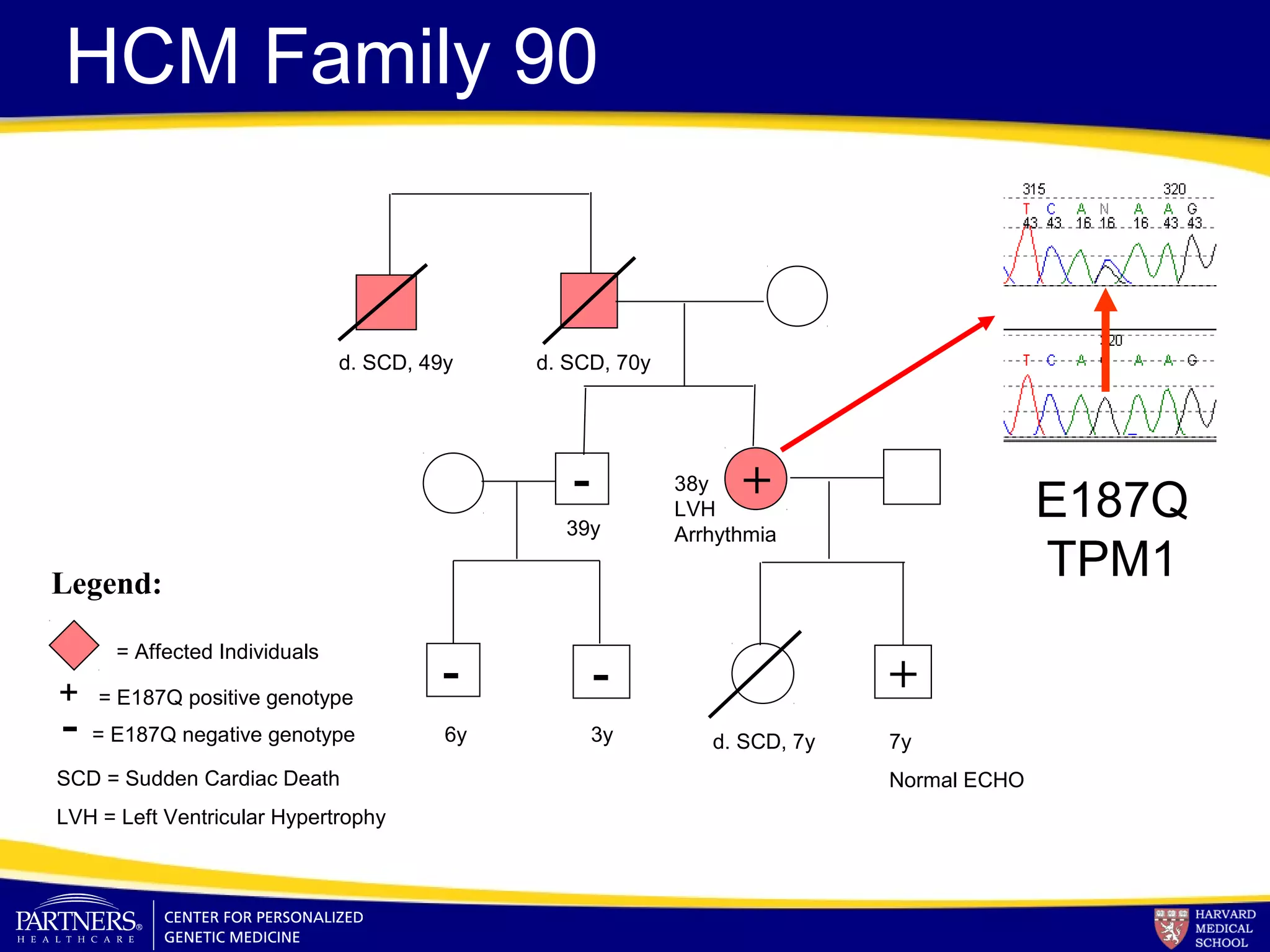





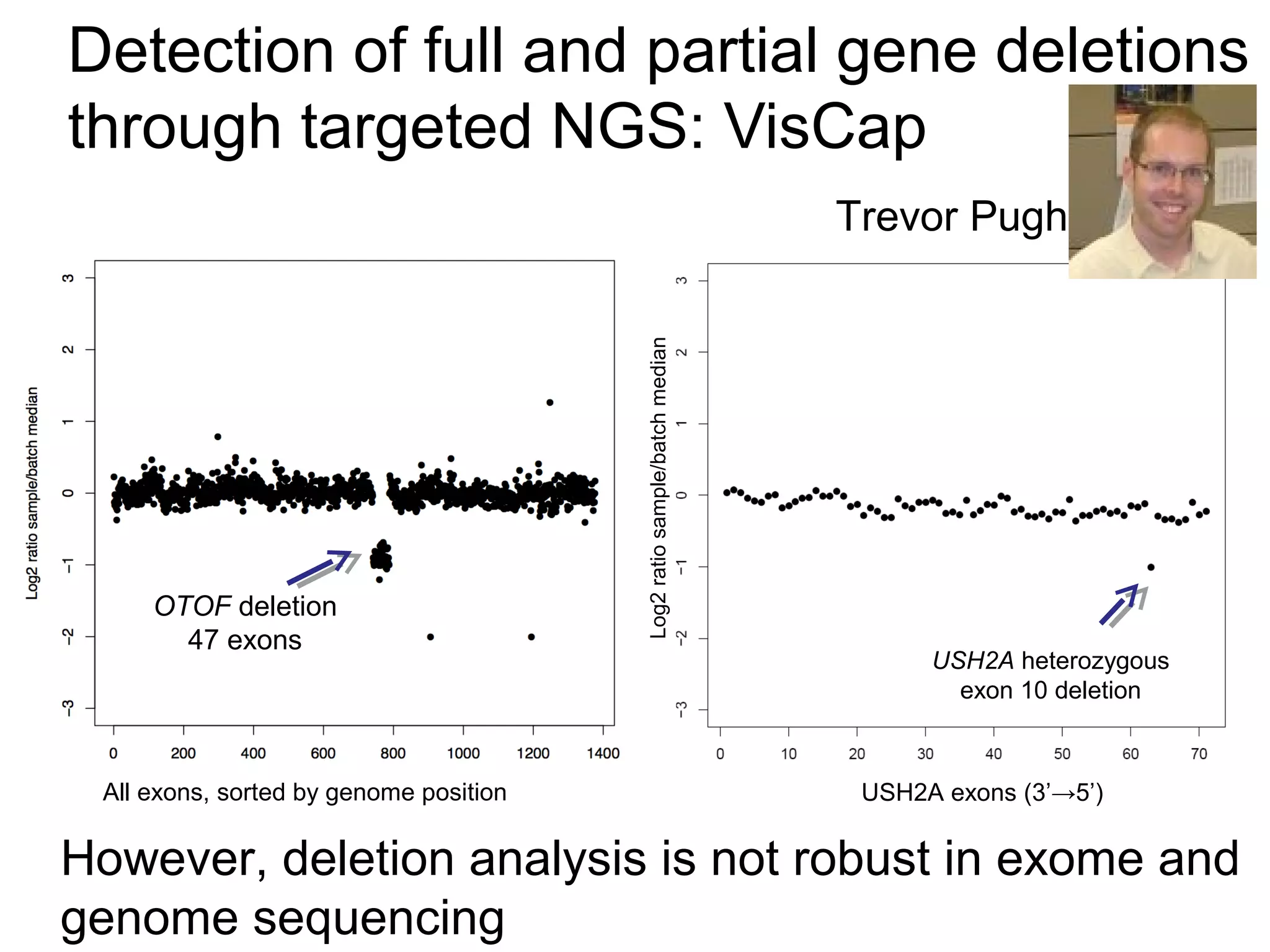
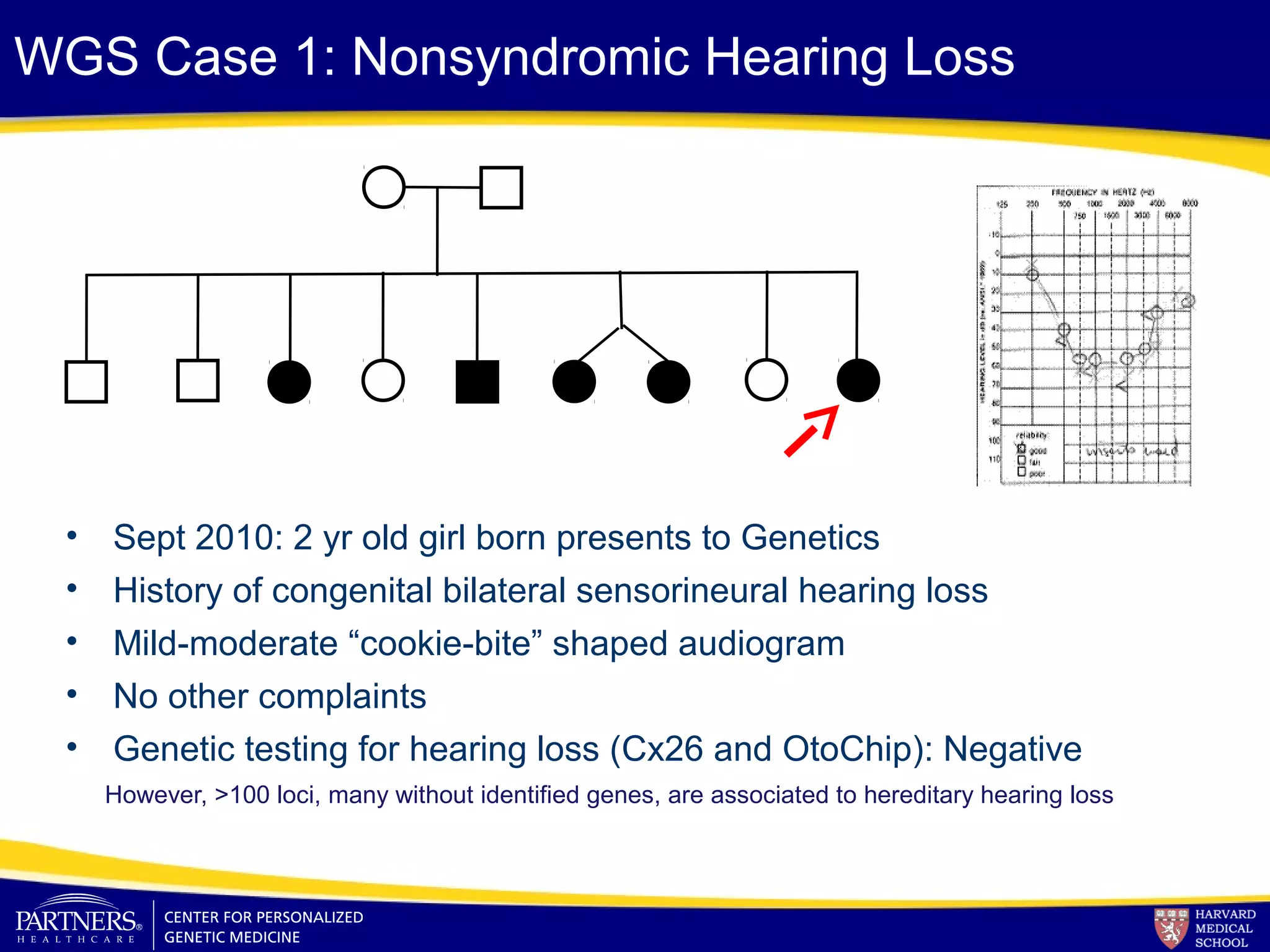
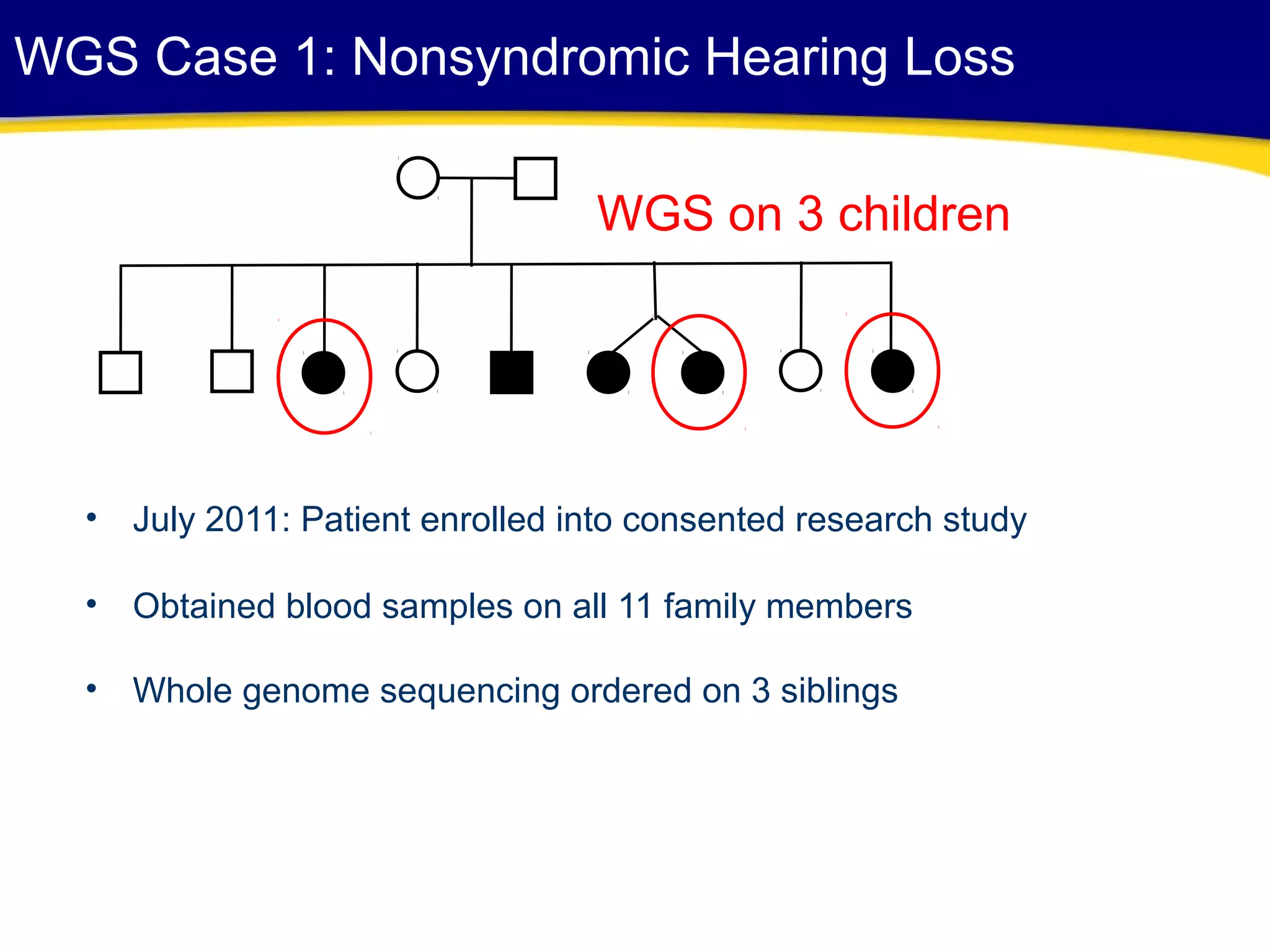




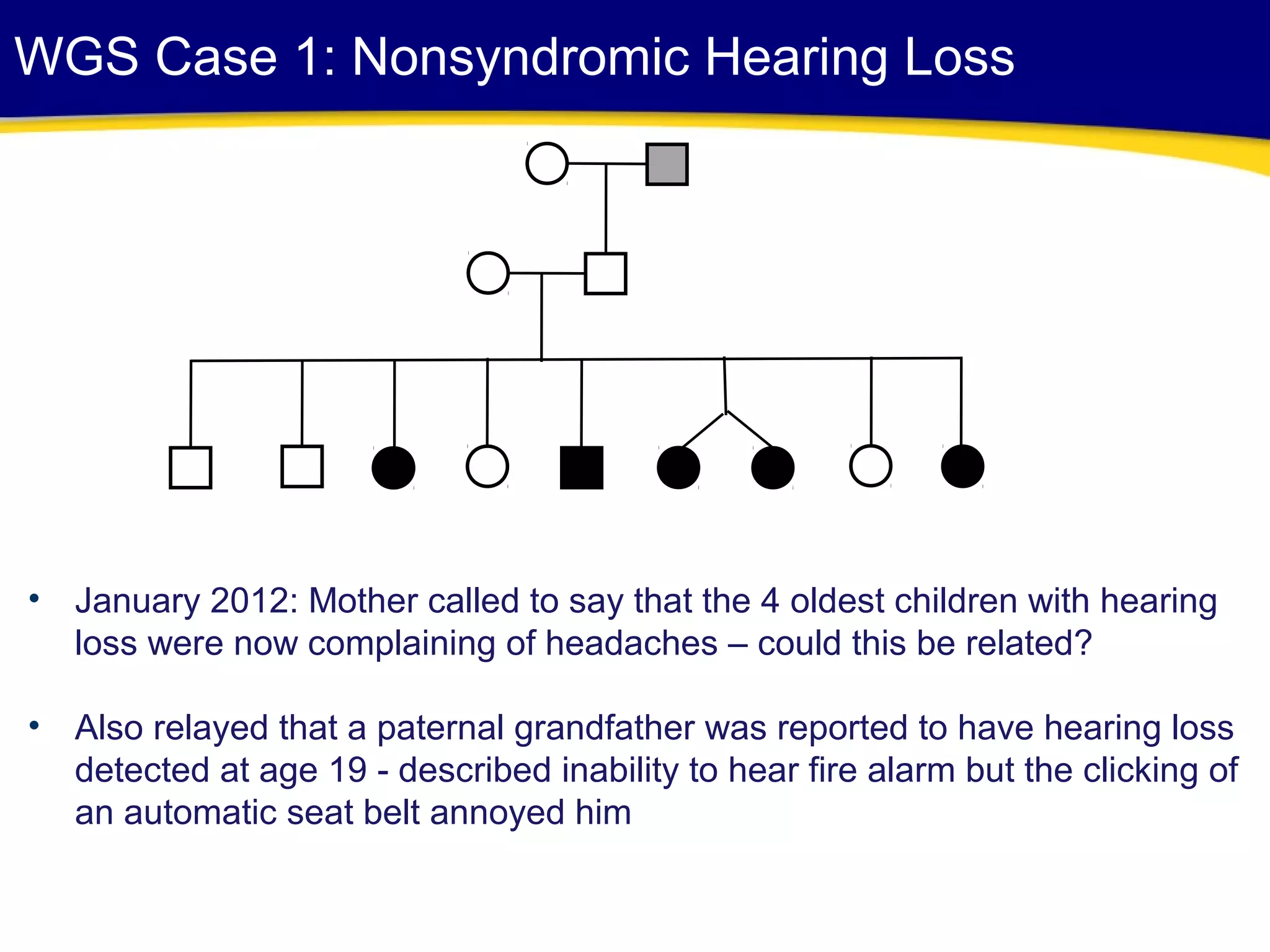


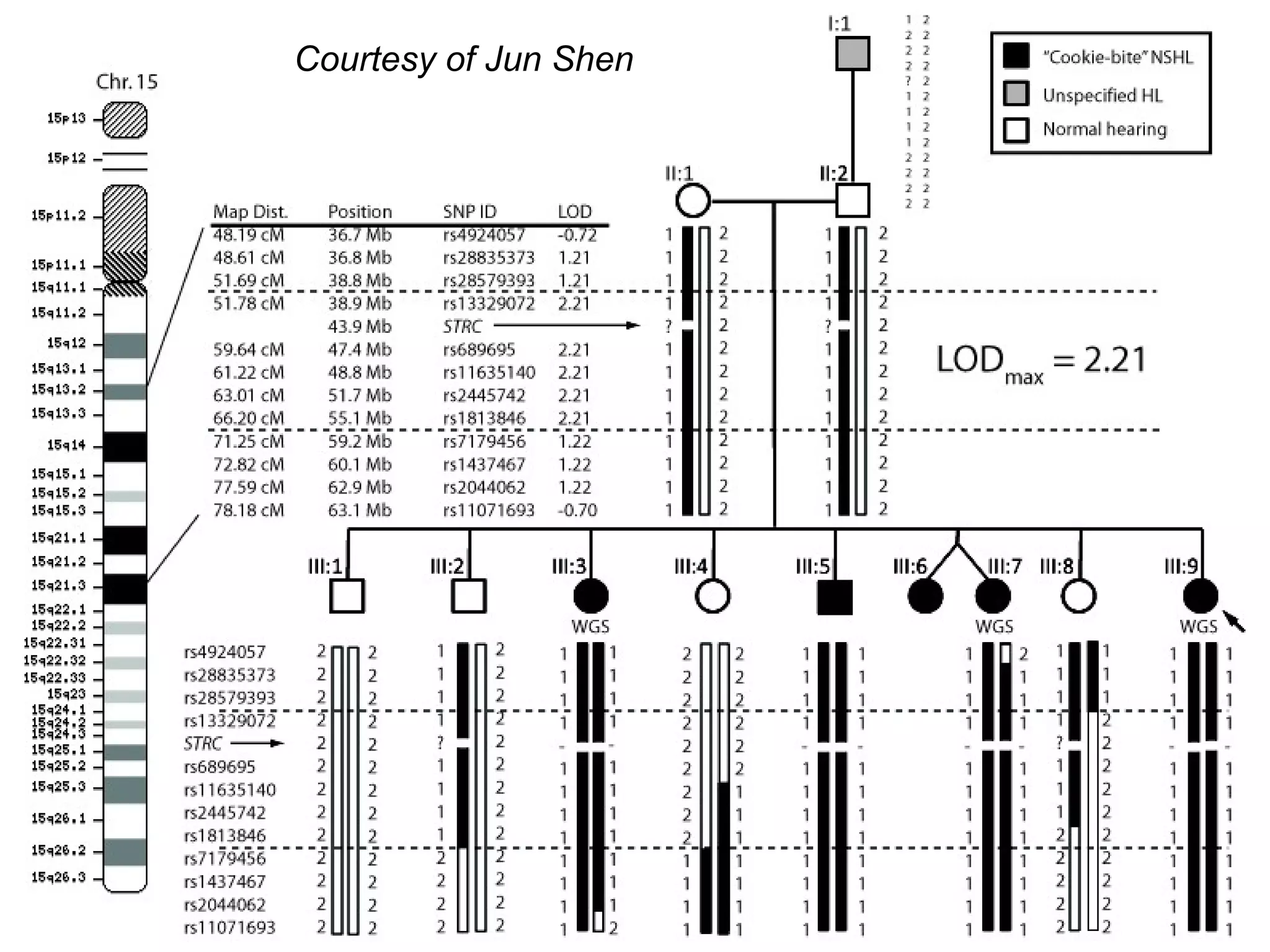
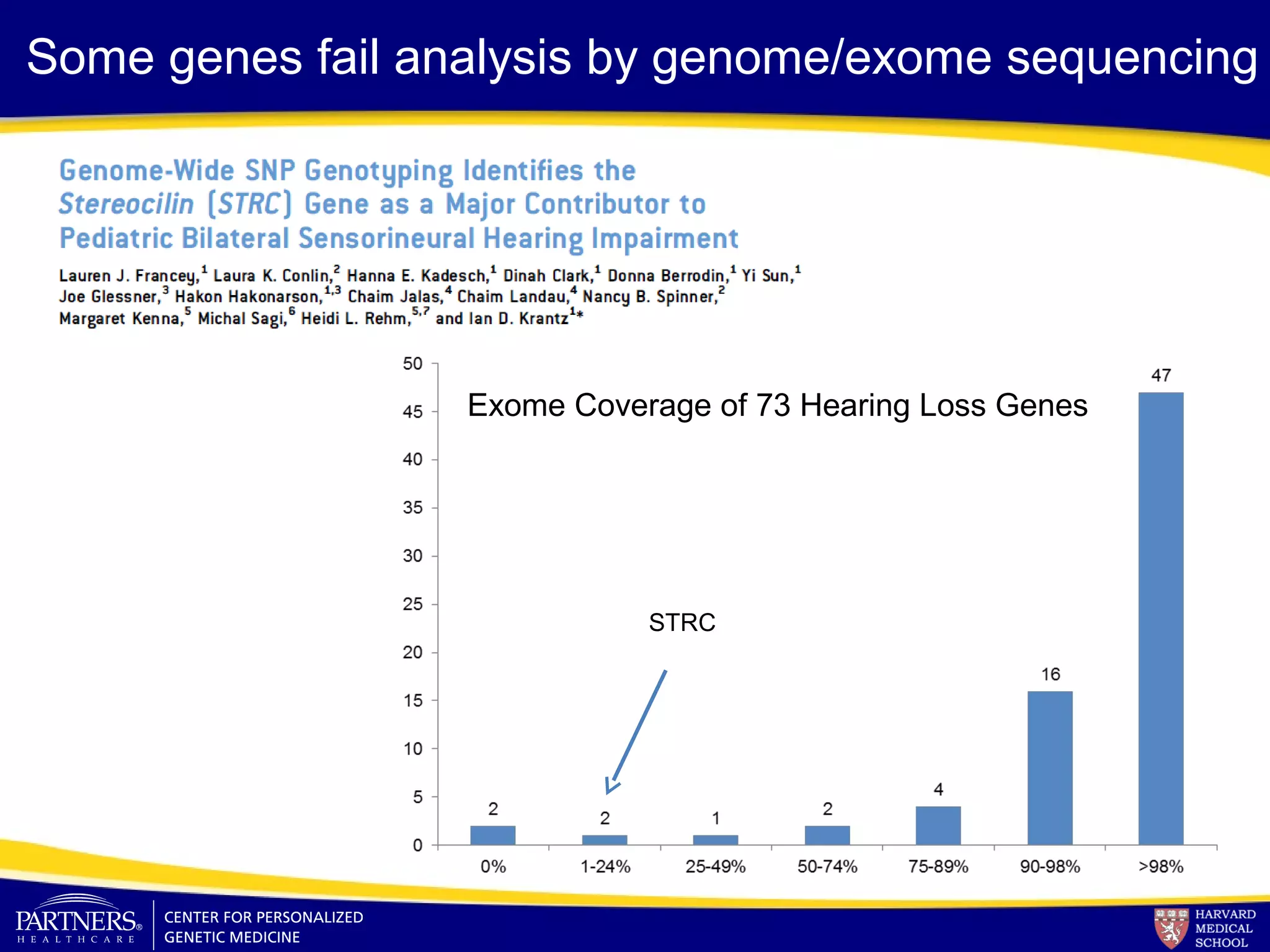
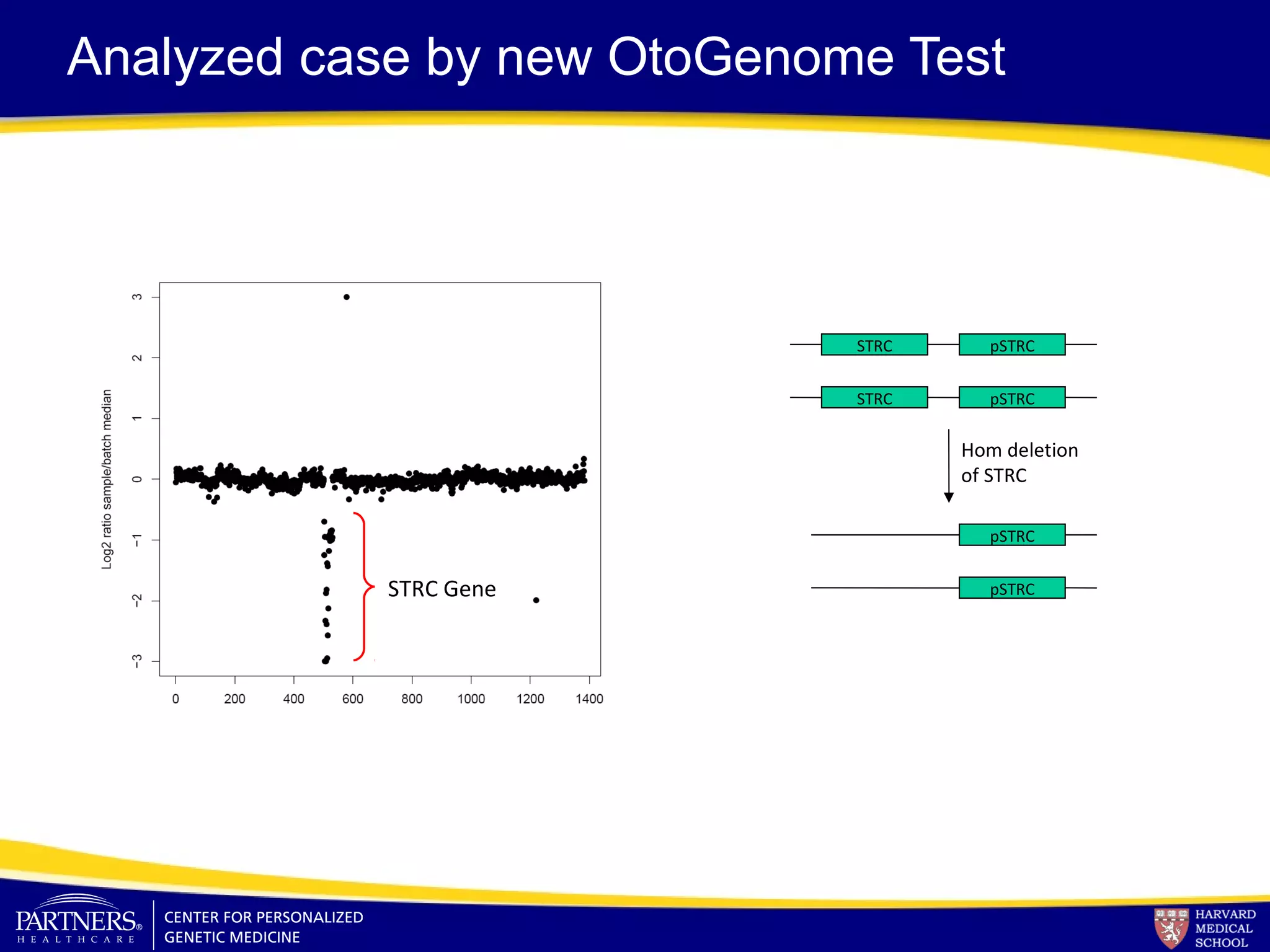

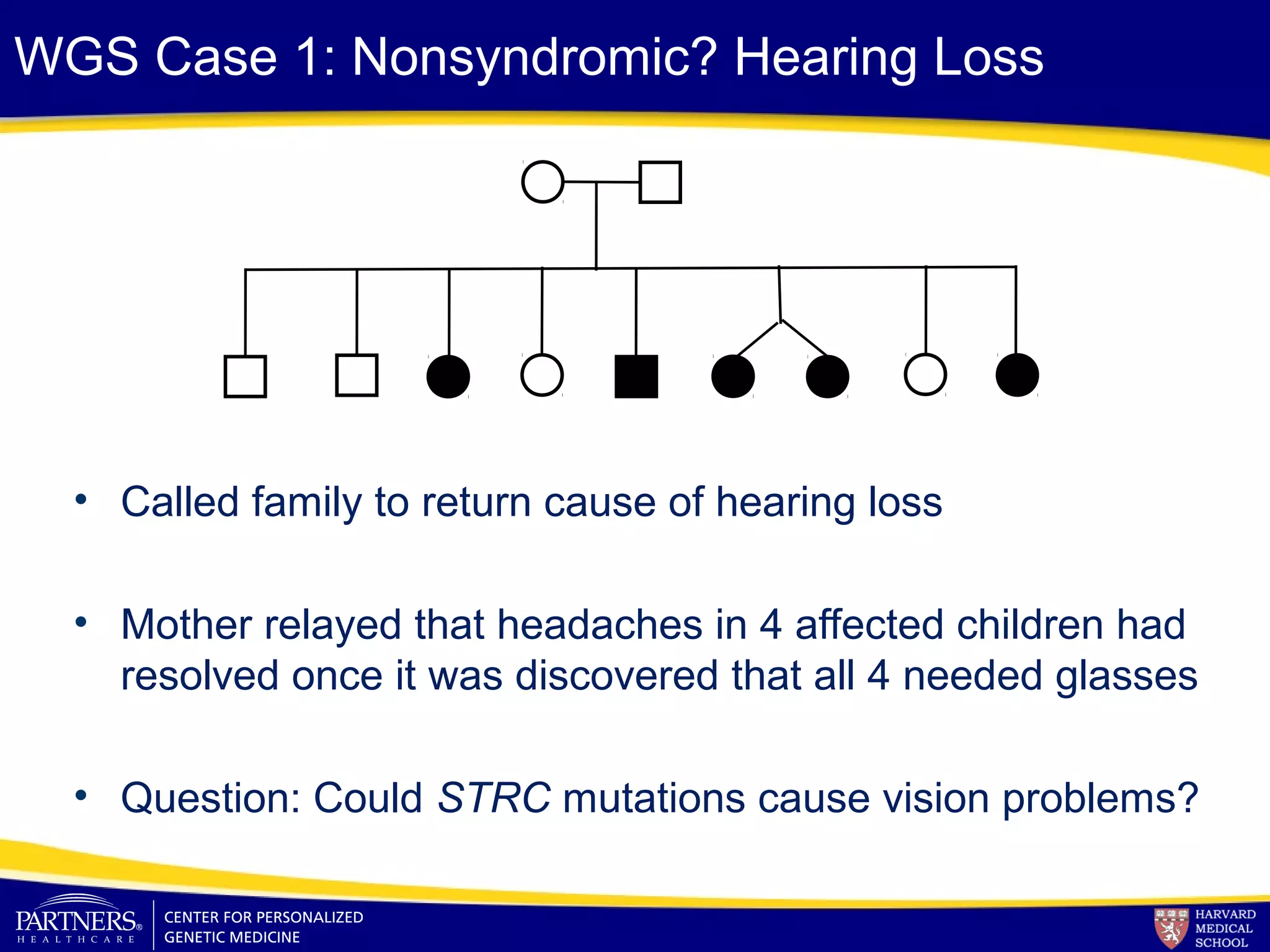
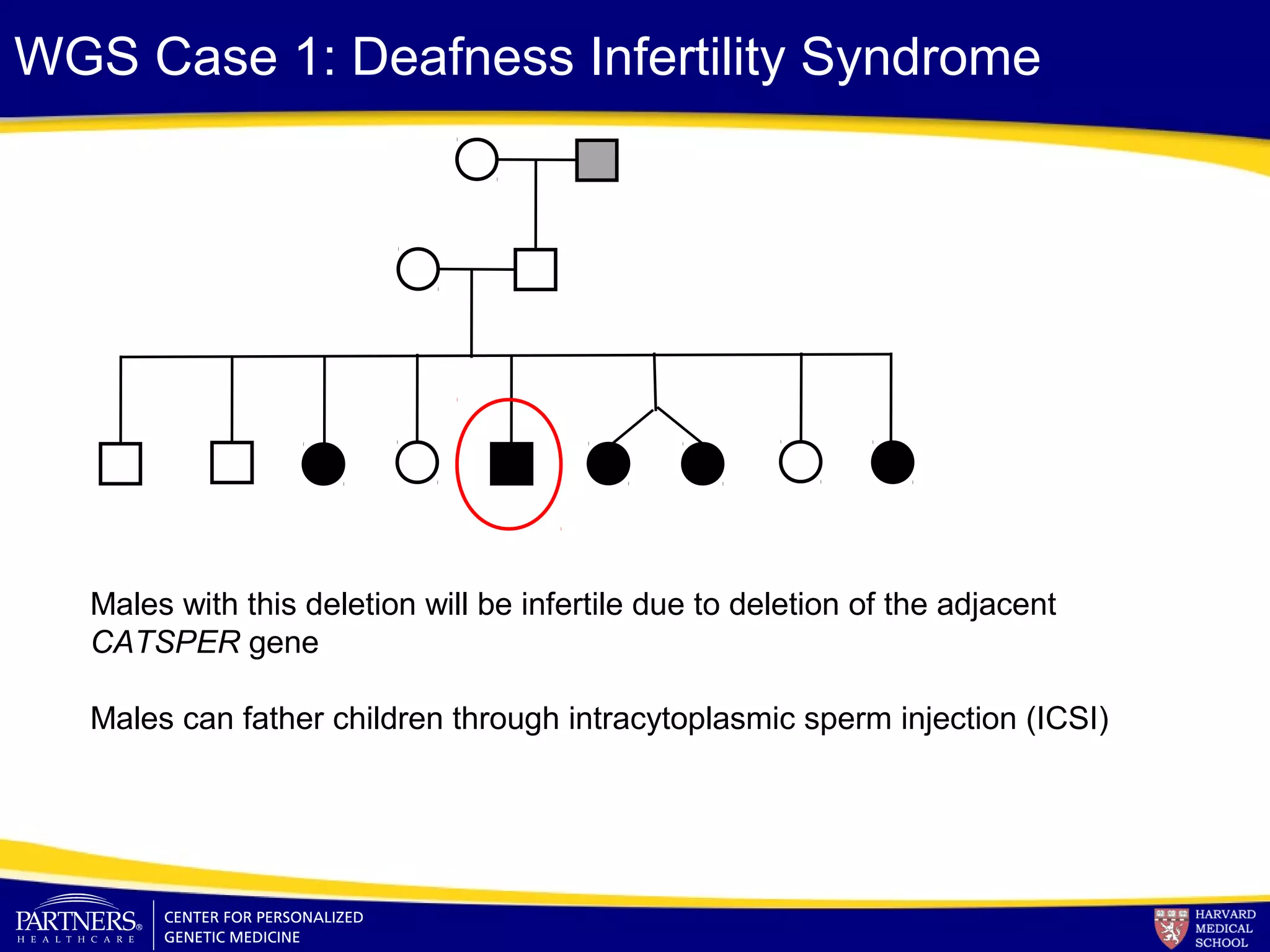




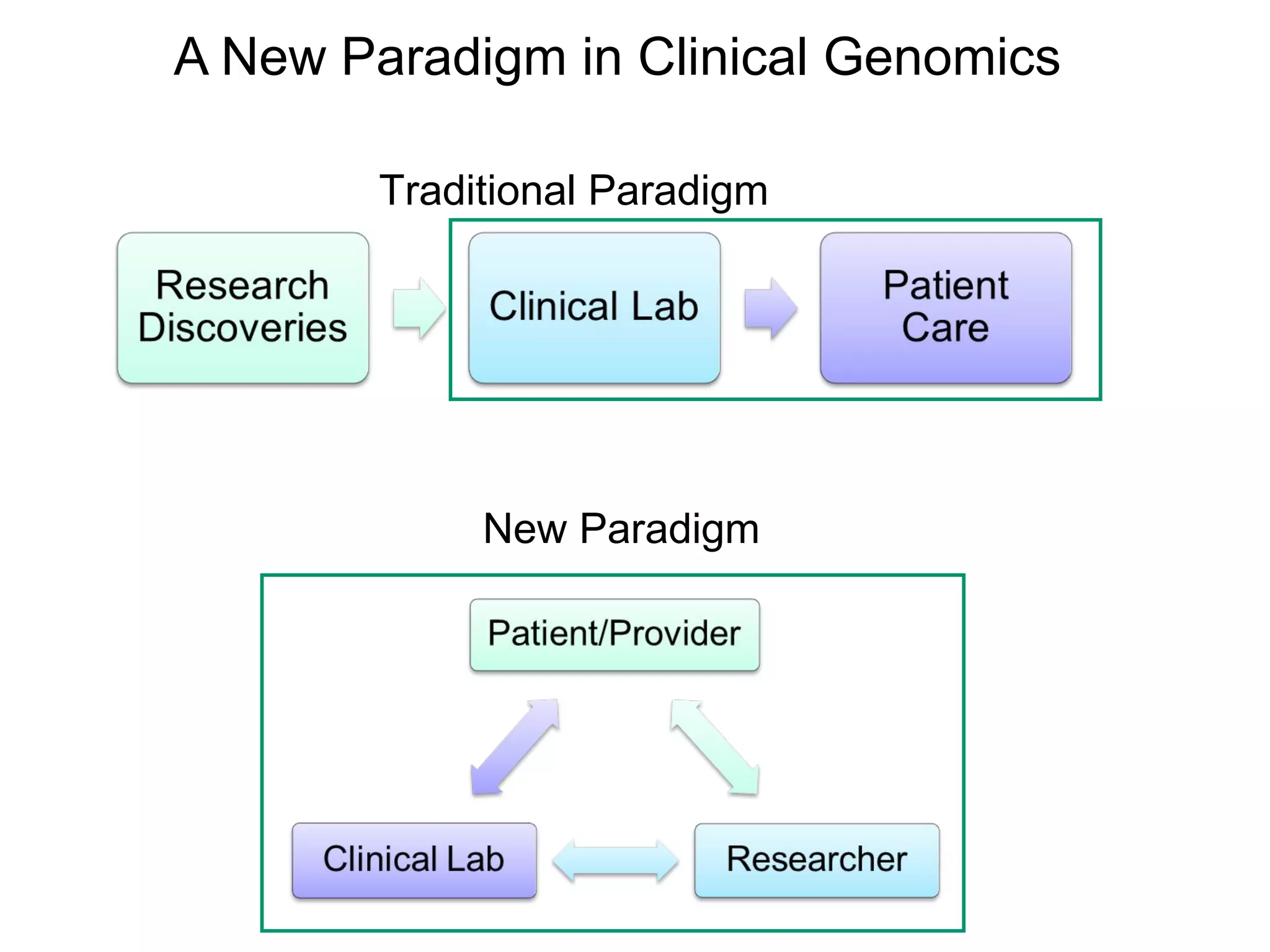

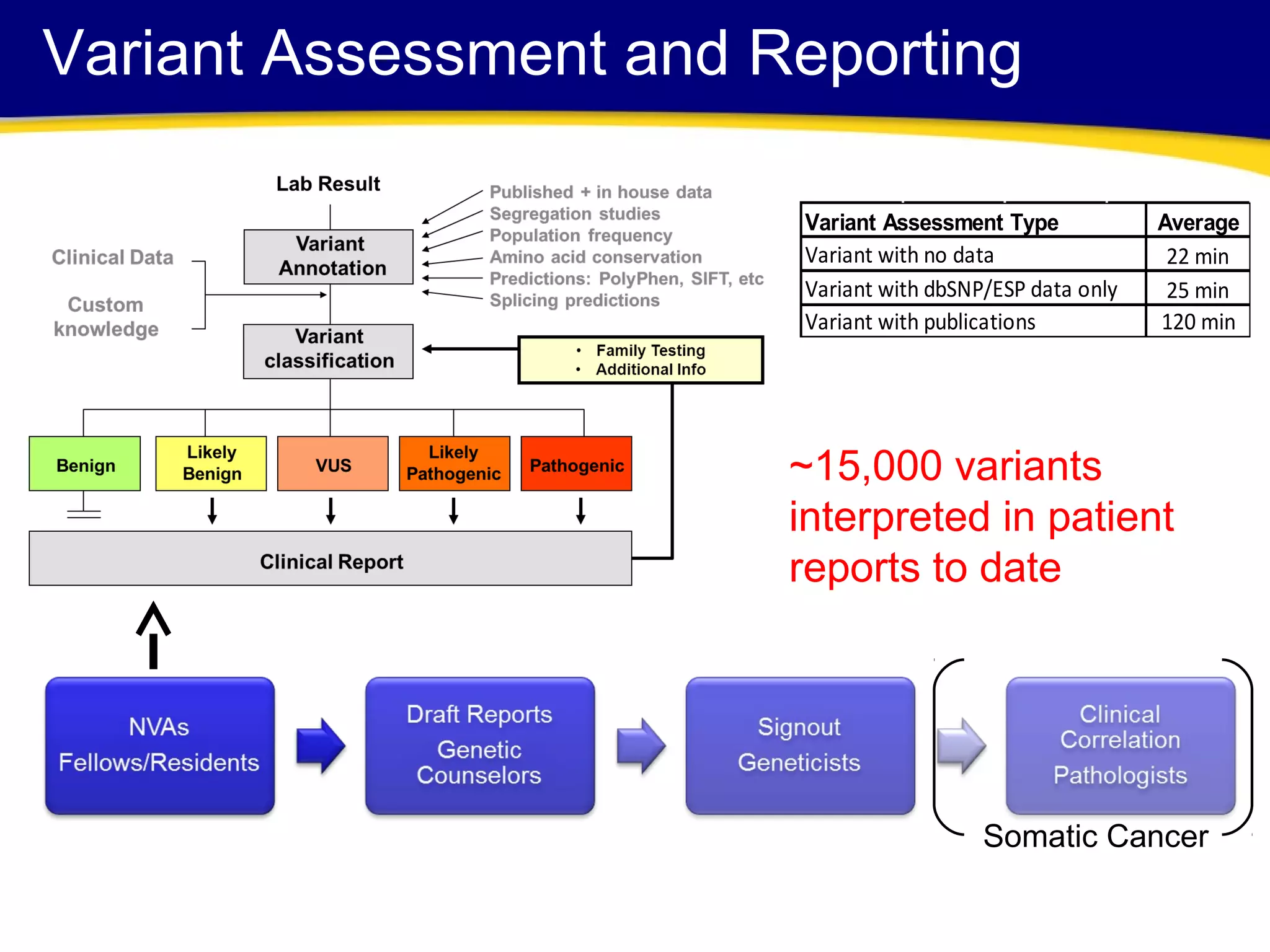

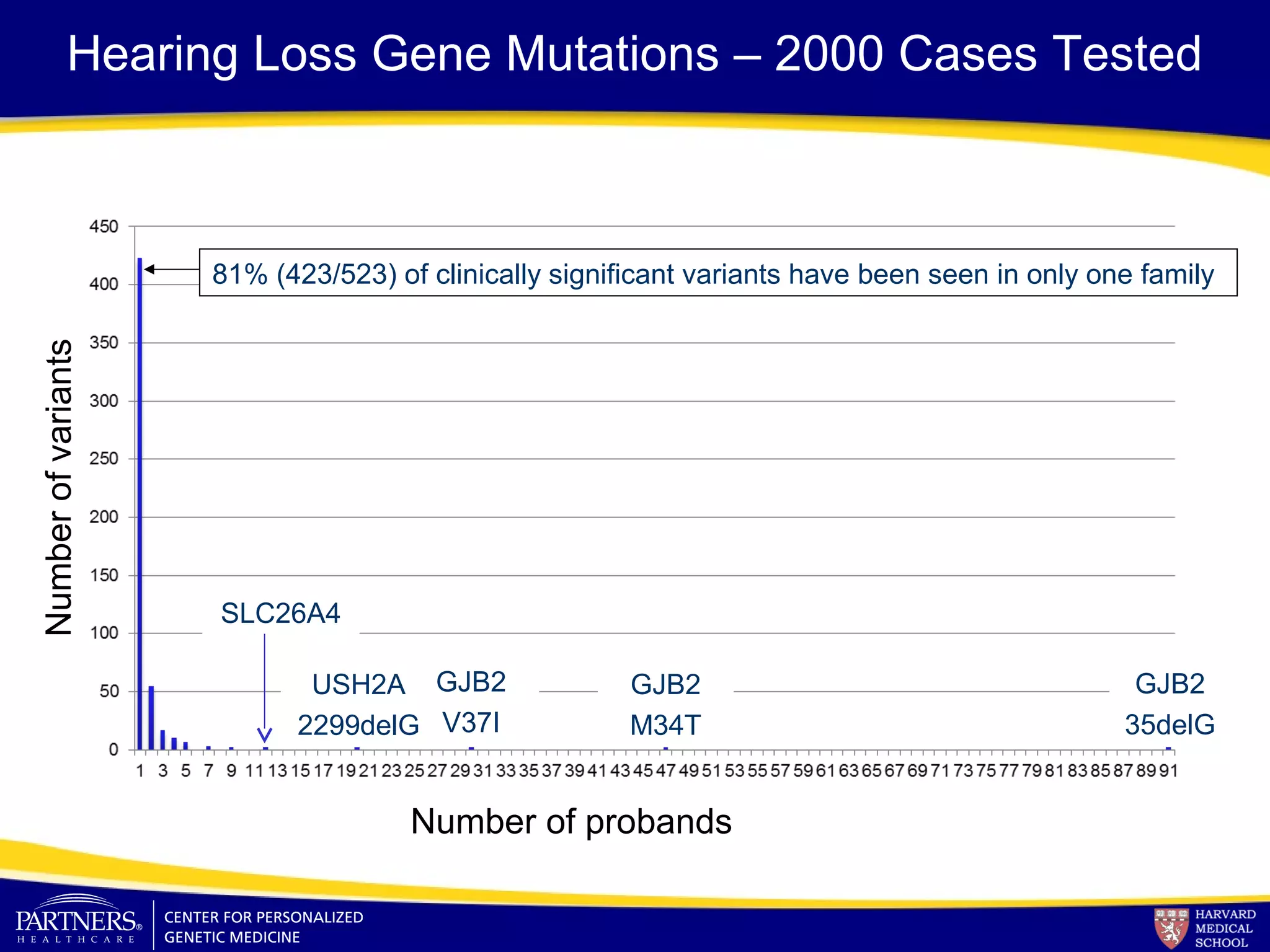
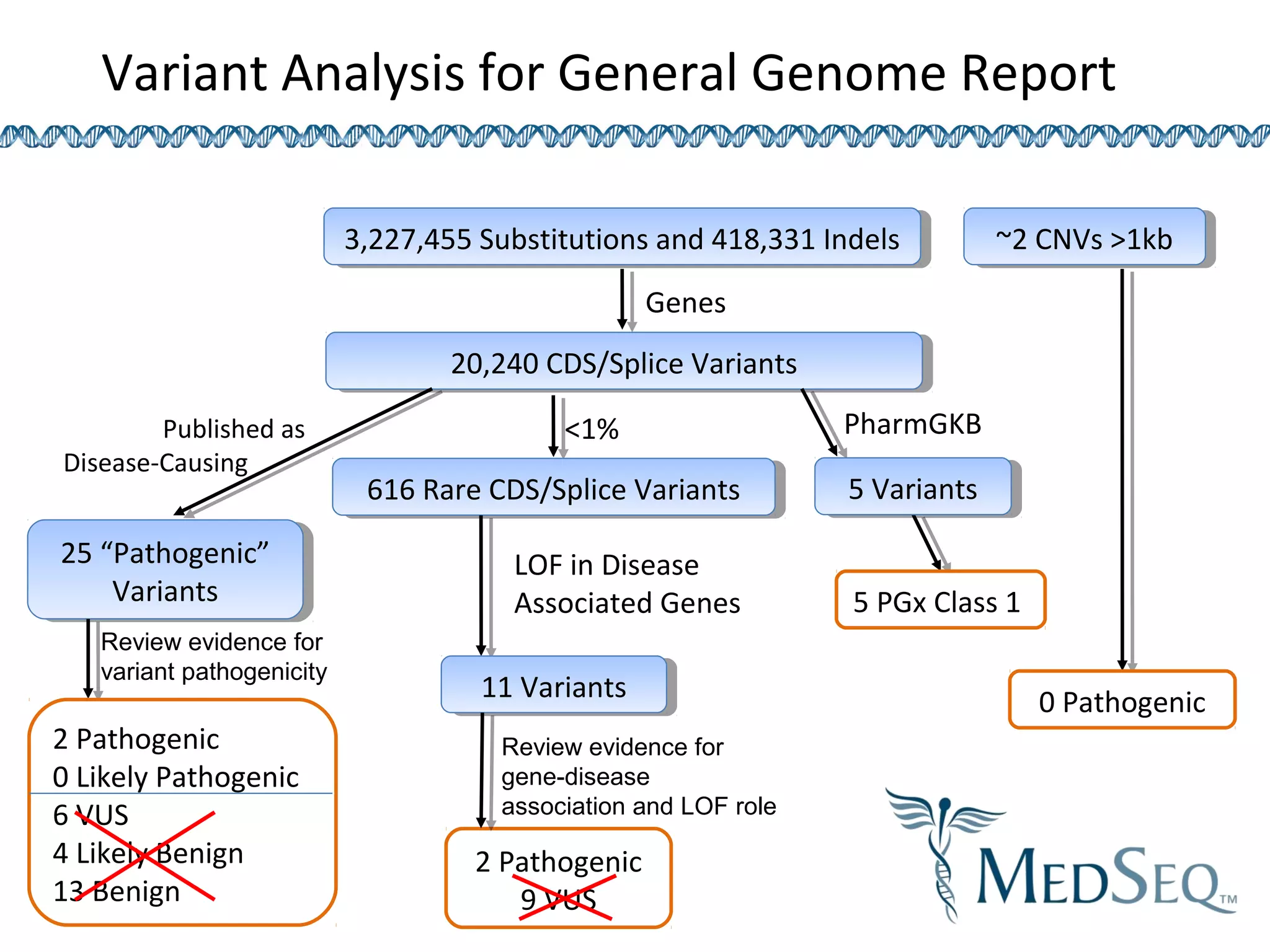
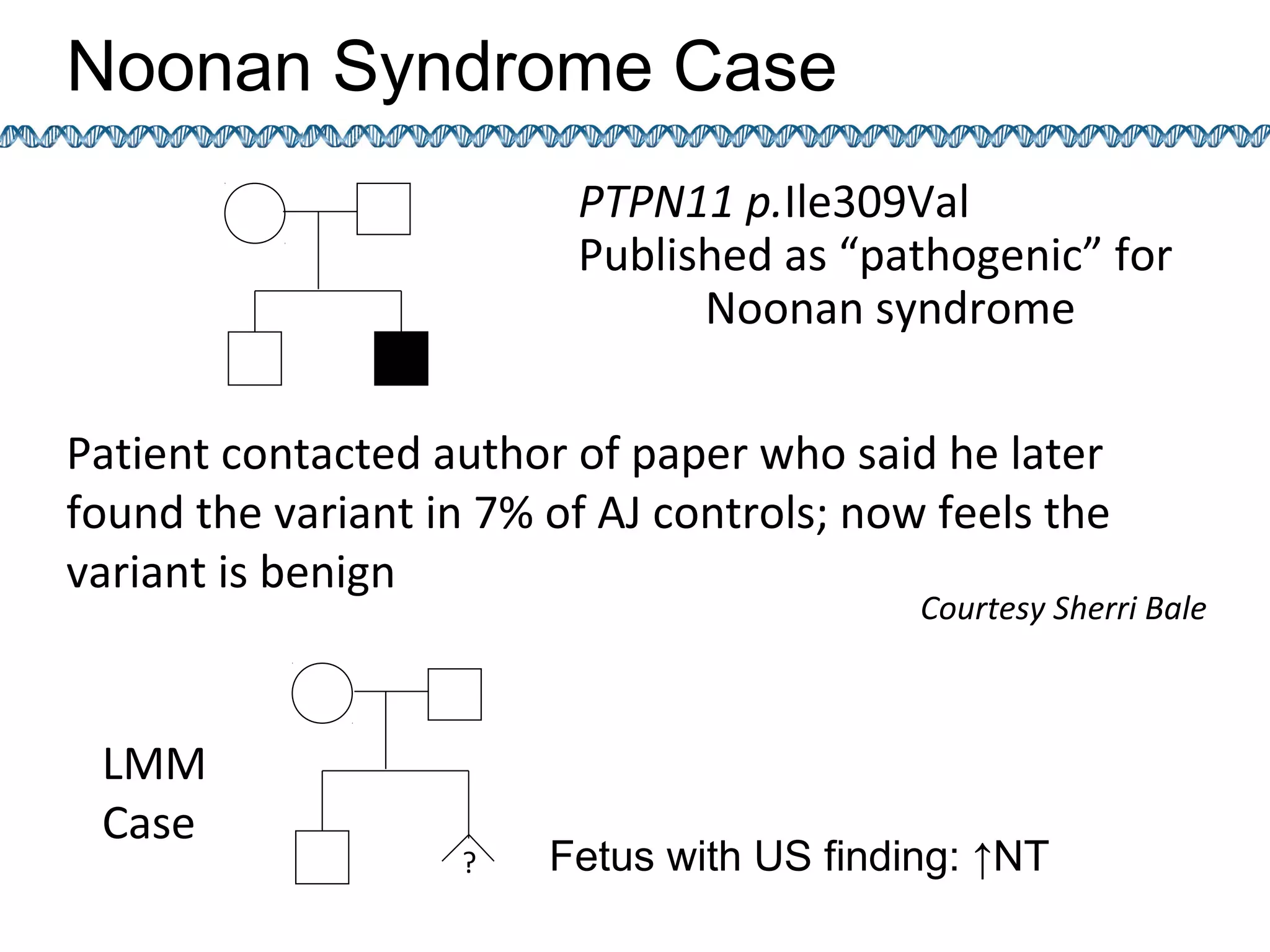



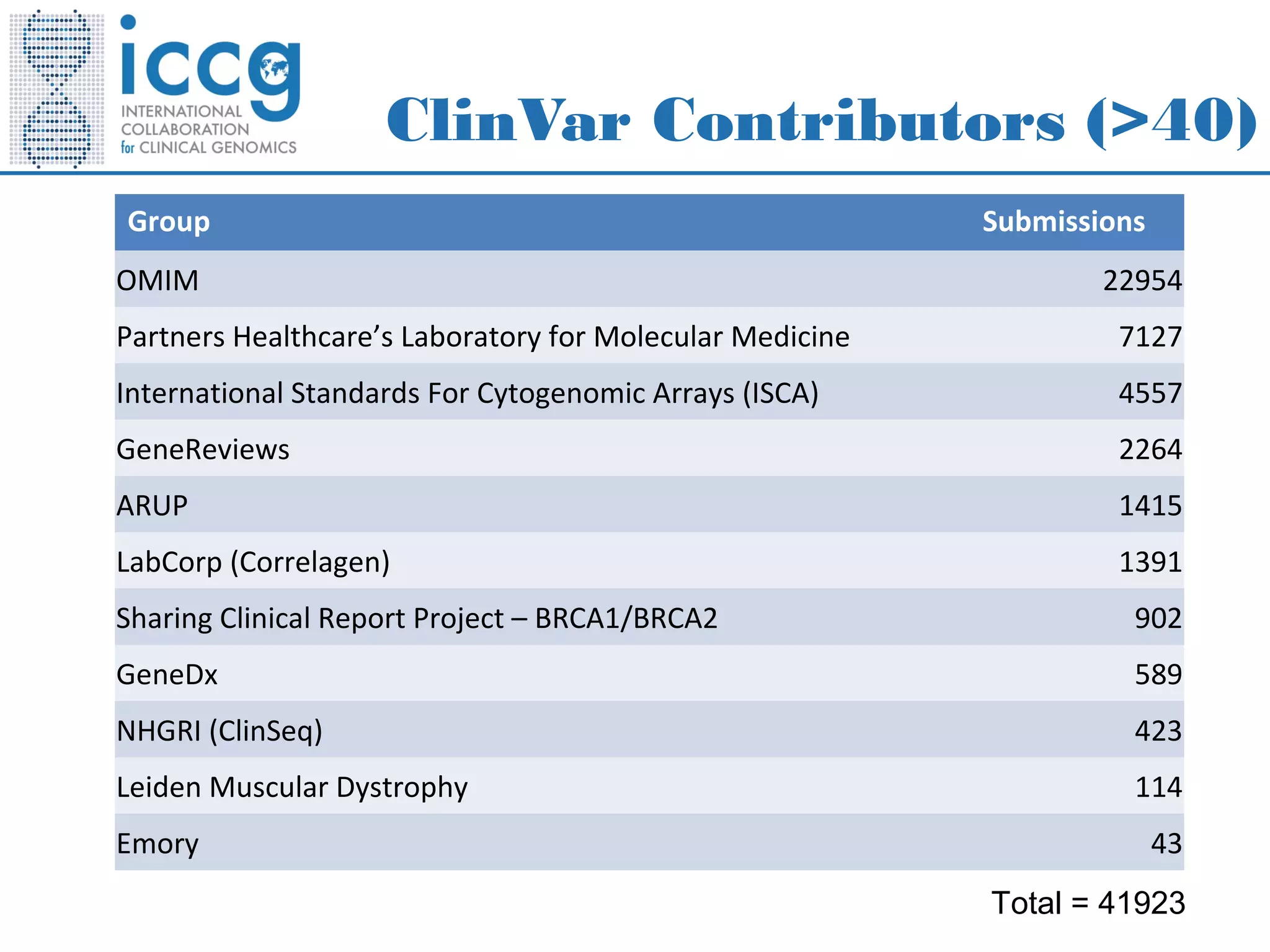
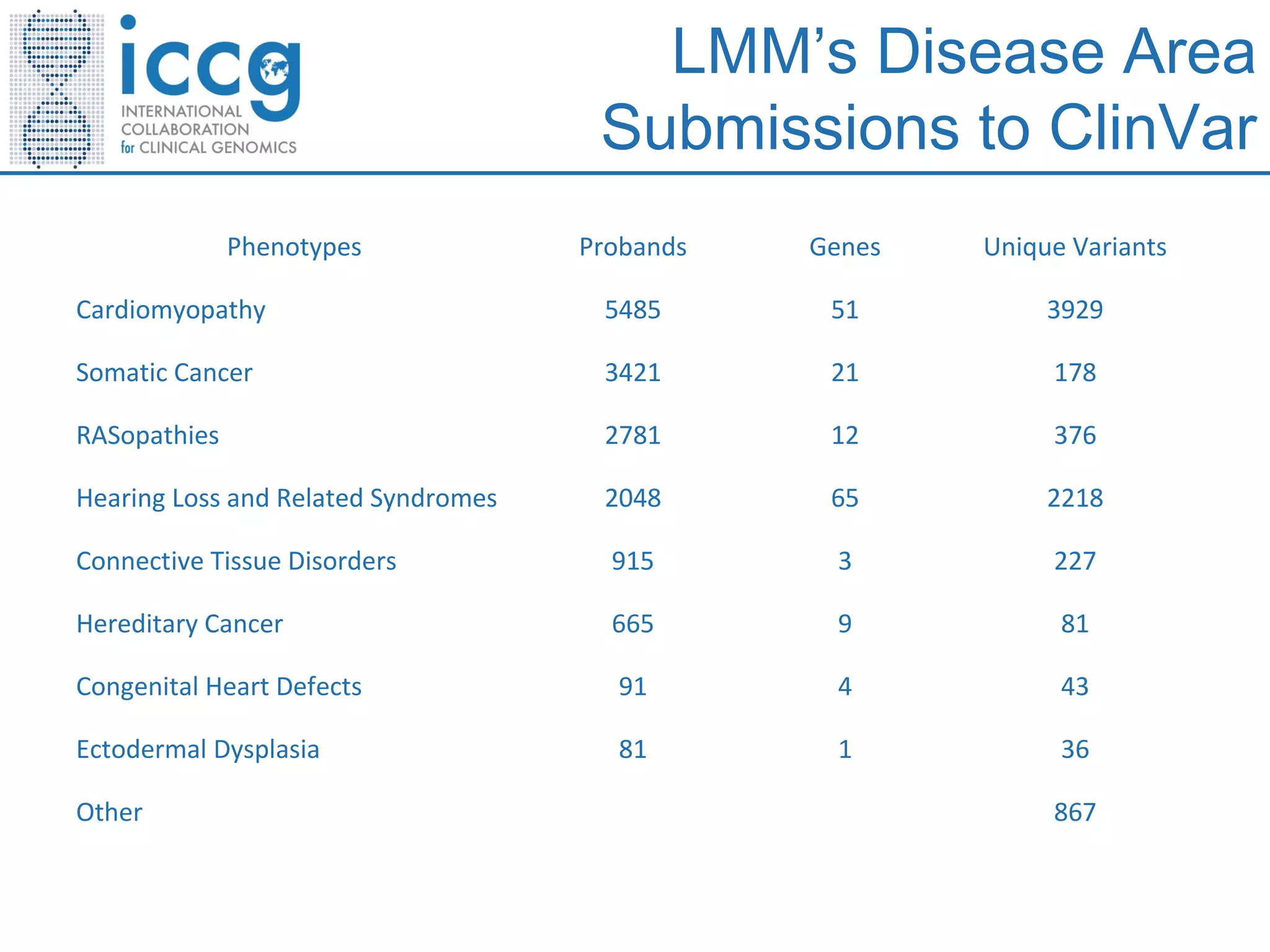




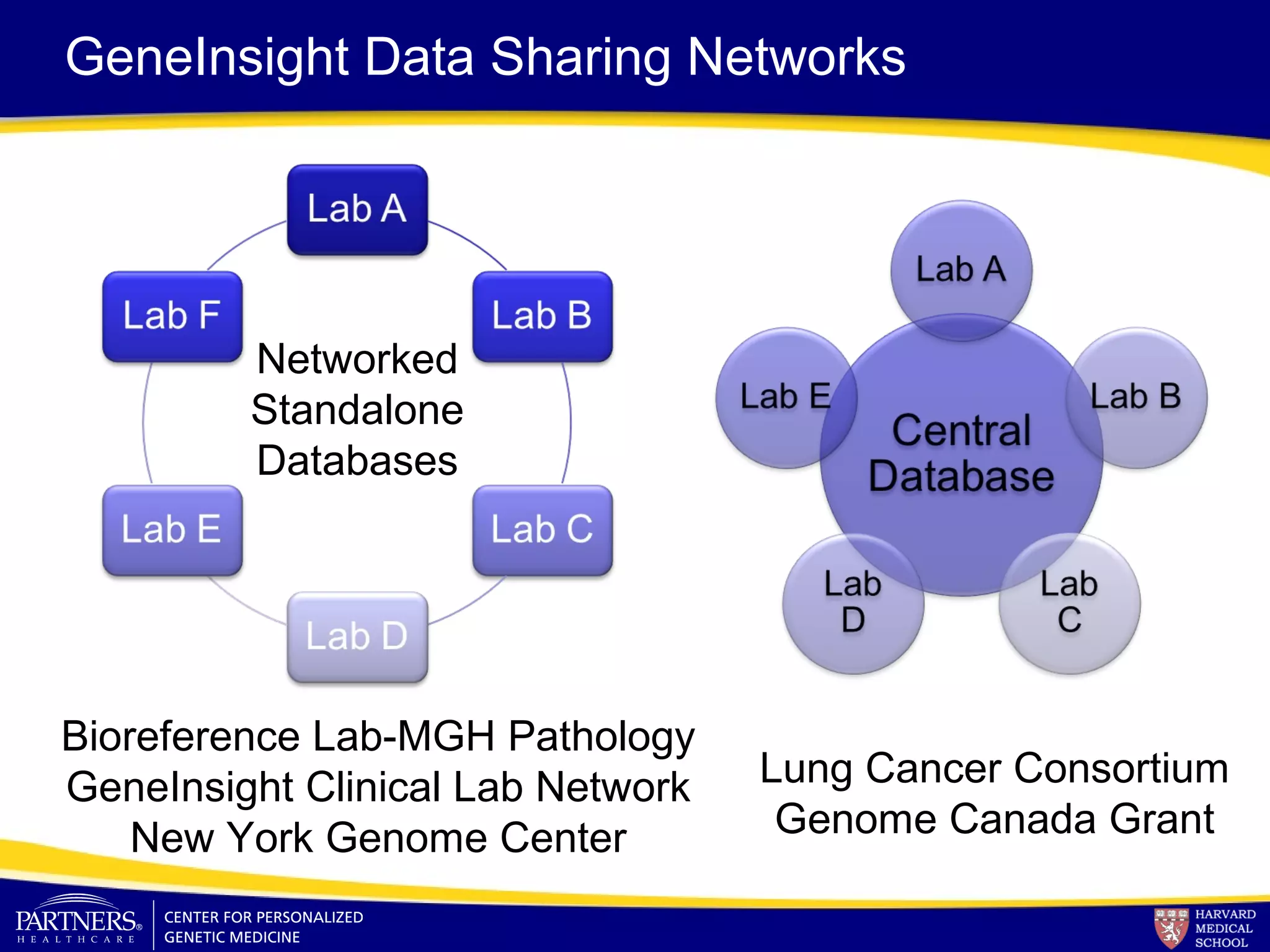
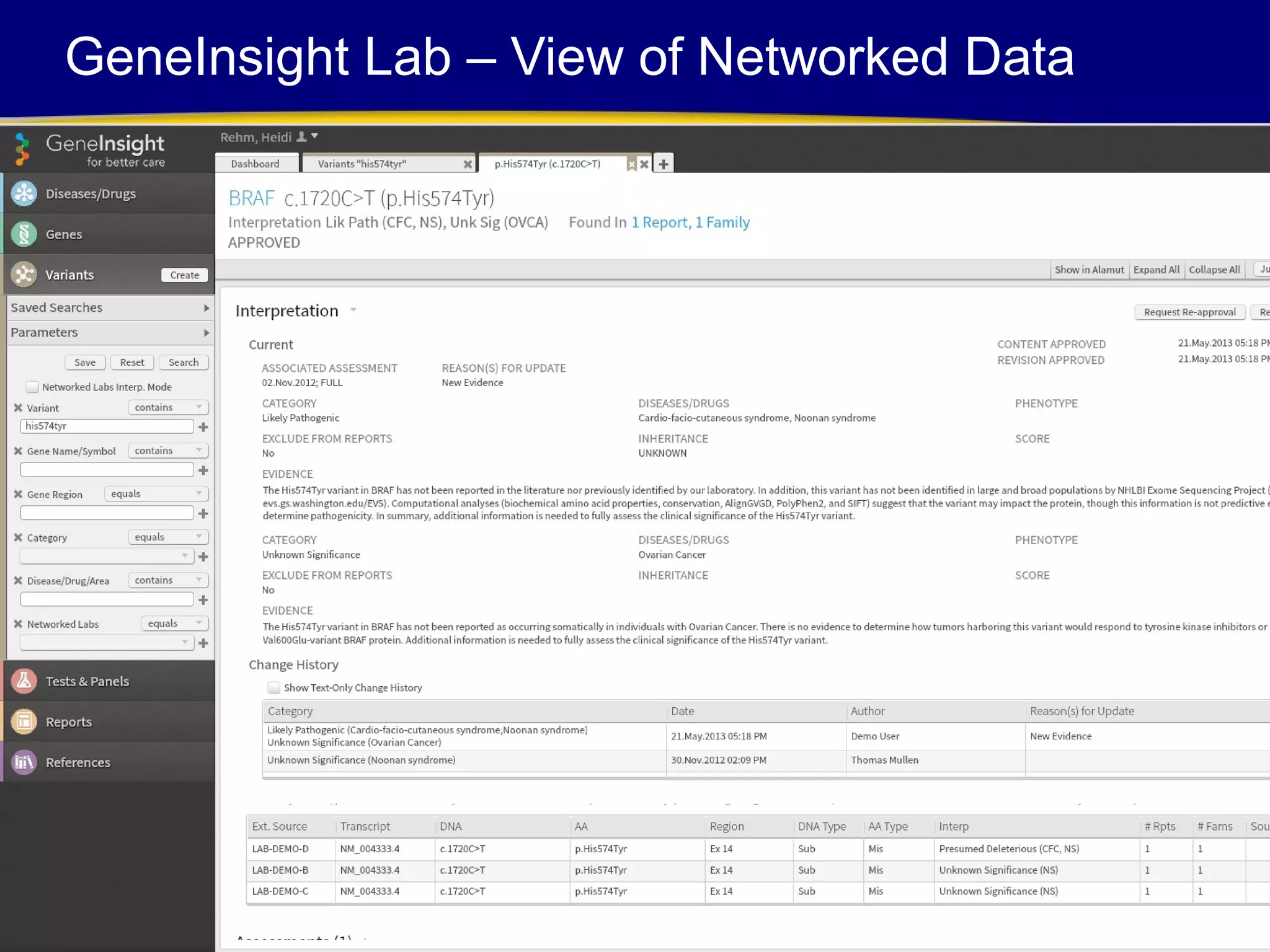
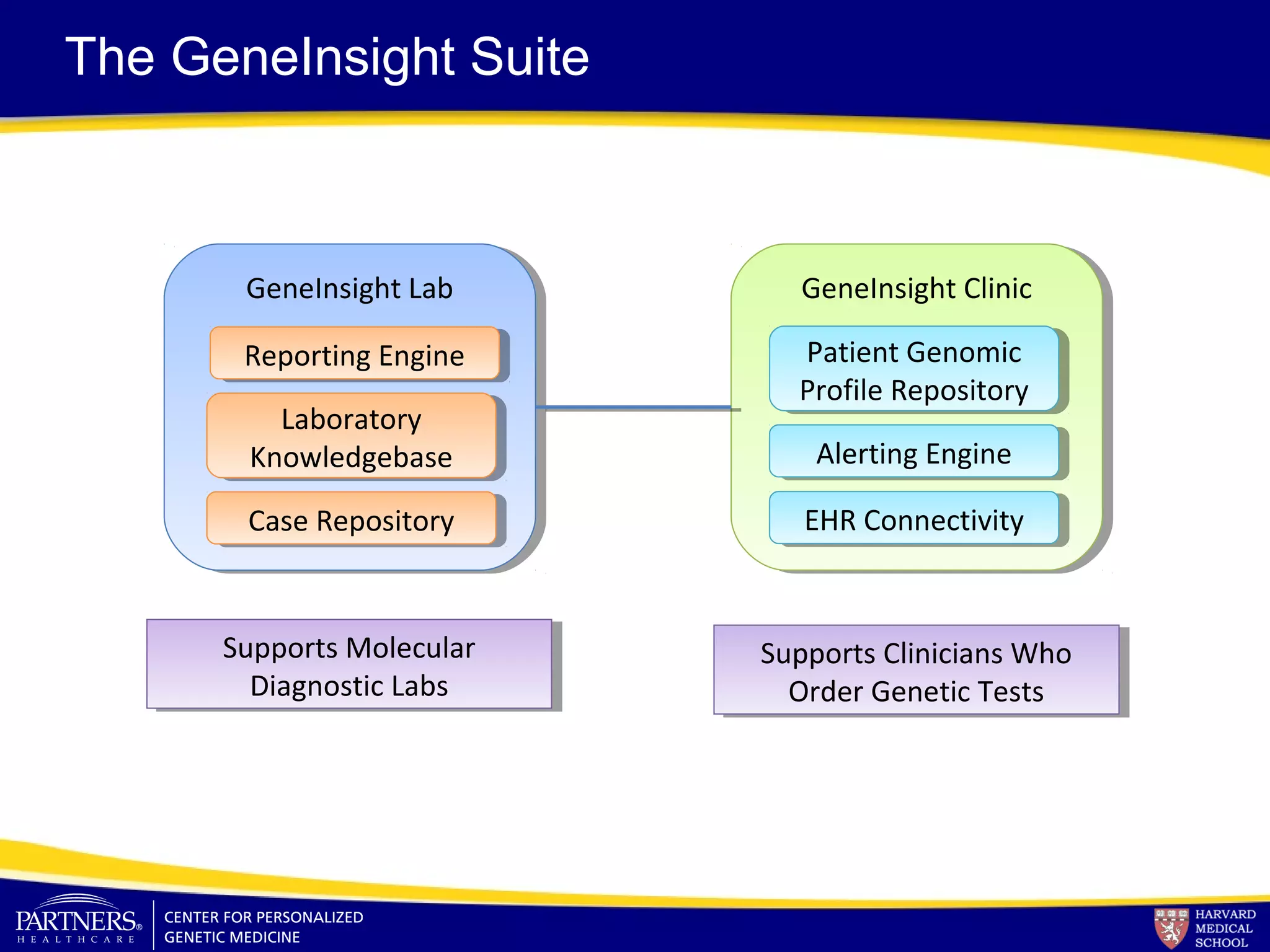
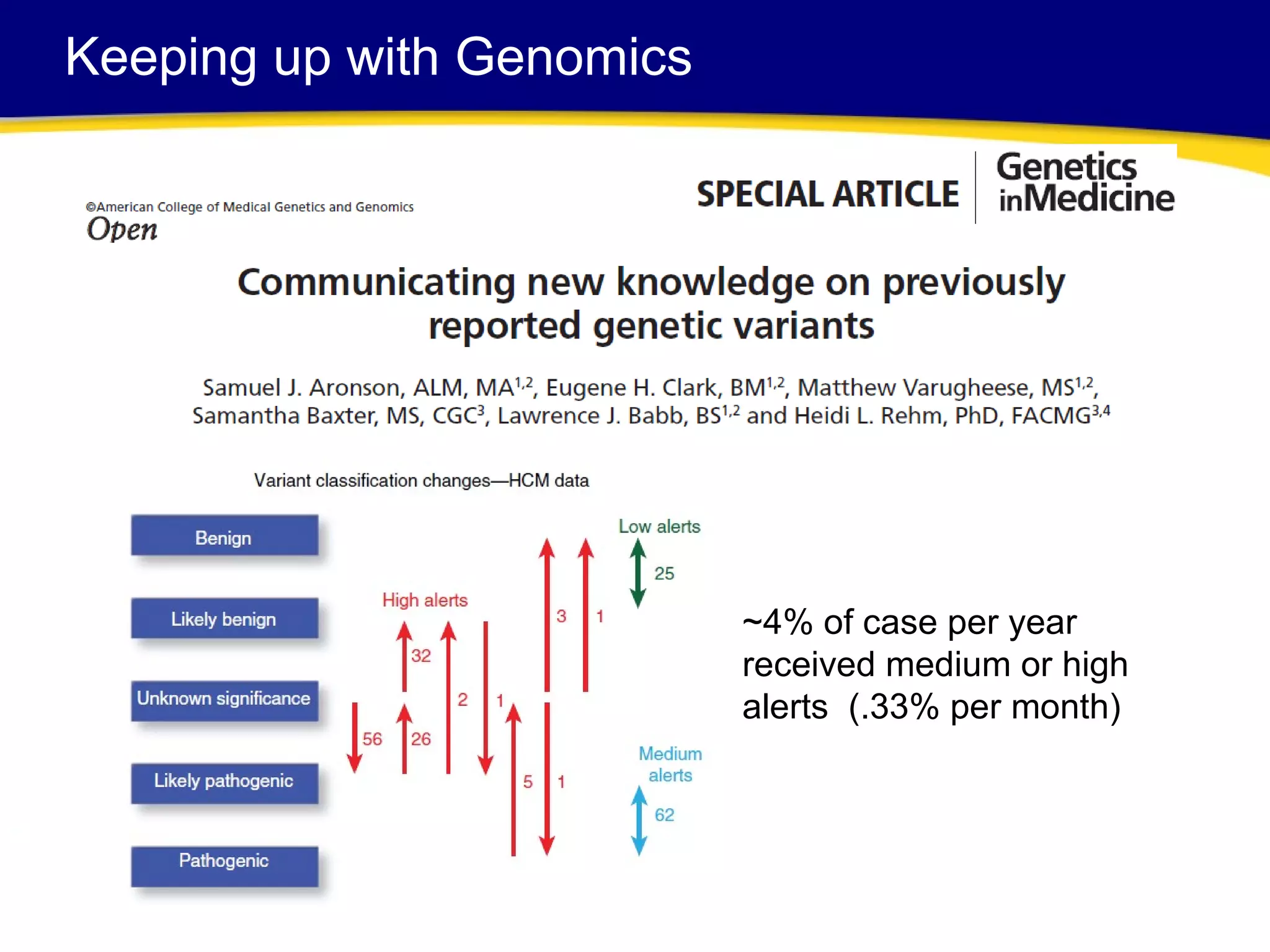
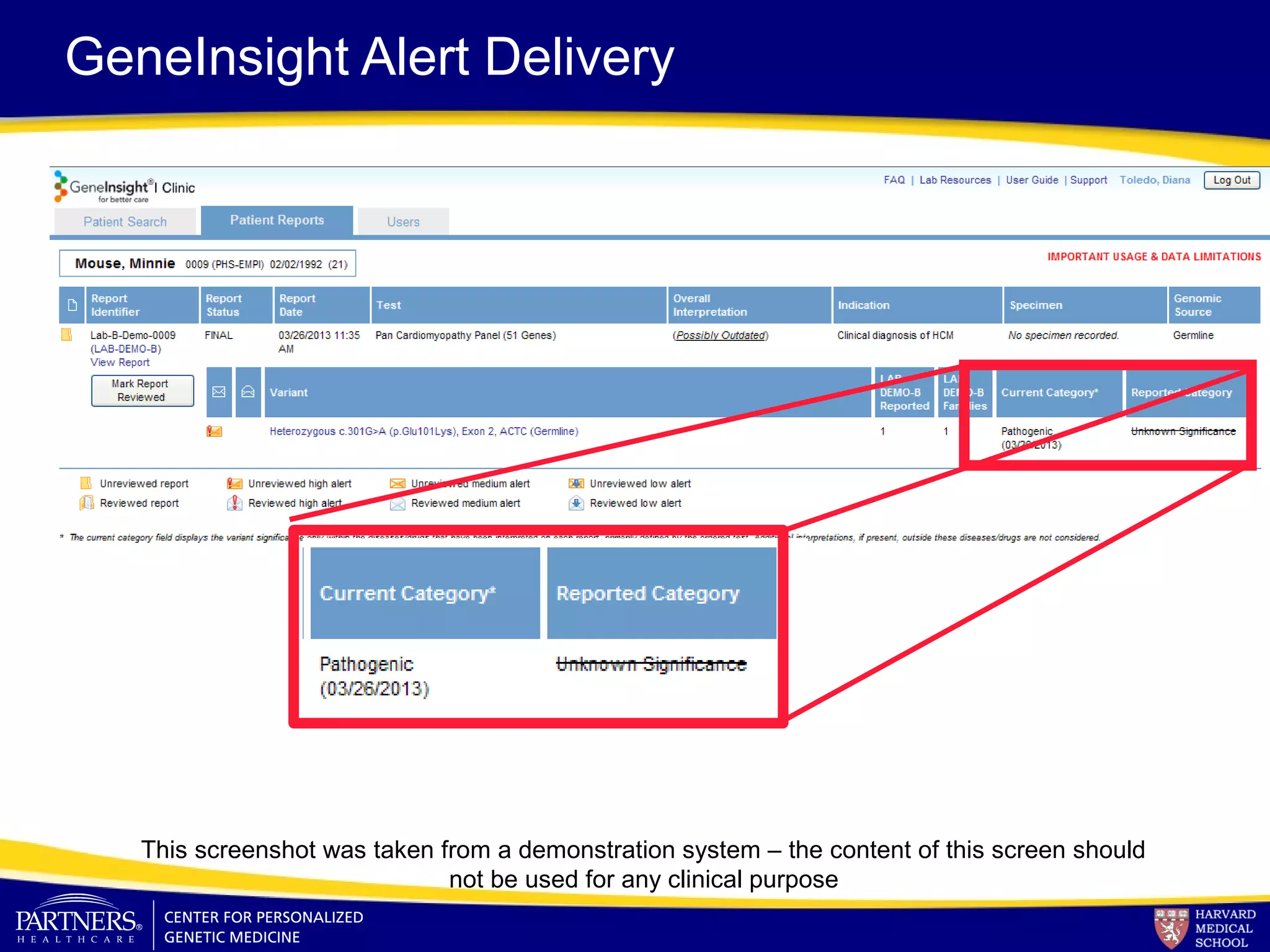

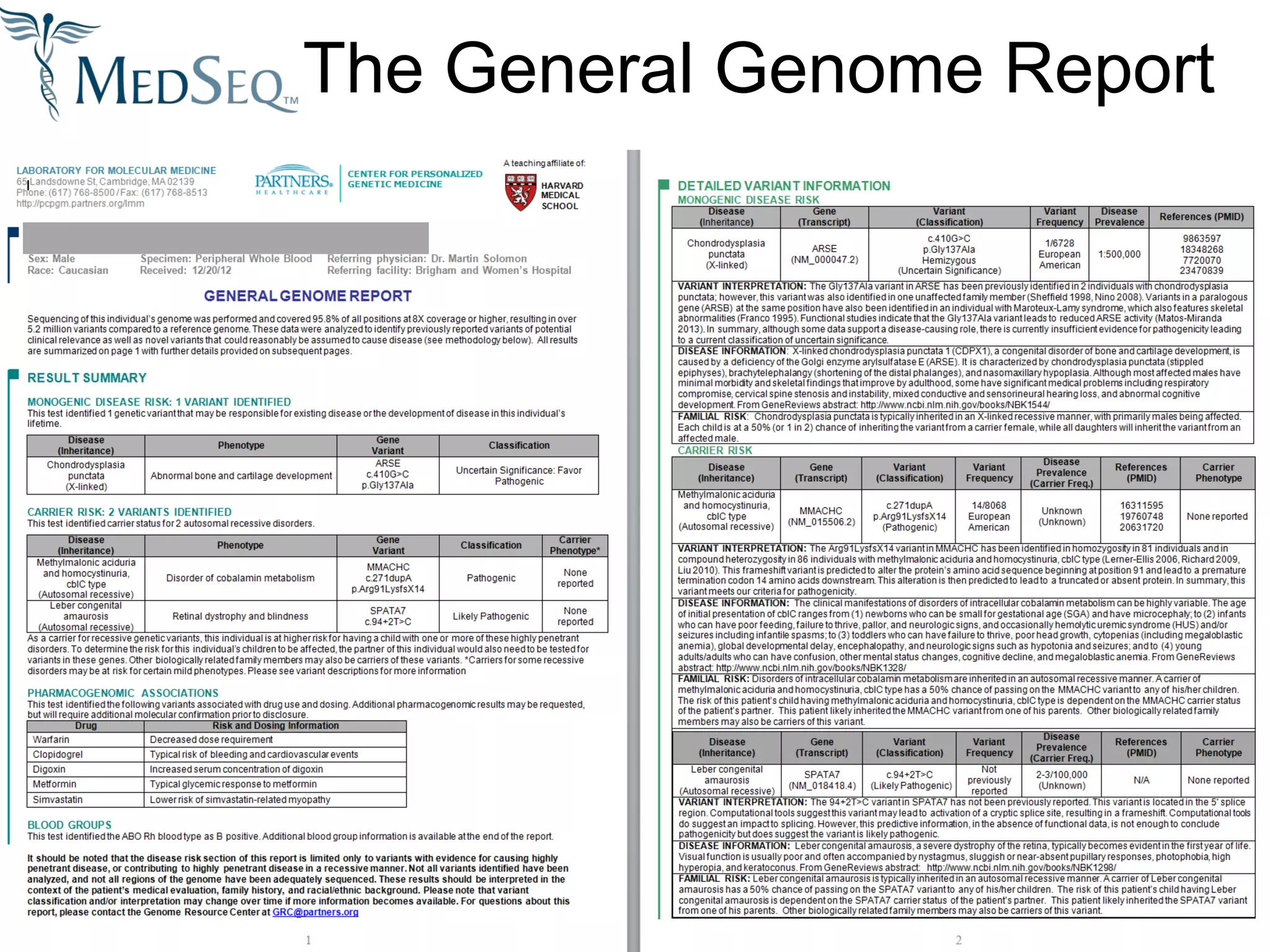

This document discusses integrating large scale genomic sequencing into clinical practice. It describes how the Laboratory for Molecular Medicine at Partners Healthcare has transitioned from testing a few genes to now offering over 150 tests using next generation sequencing technologies. It also discusses the evolution of testing panels, moving from targeted panels to exome and genome sequencing. Validation of sequencing methods and challenges of variant interpretation are discussed.


























































































![Coded Agents – with UiPath SDK + LangGraph [Virtual Hands-on Workshop]](https://cdn.slidesharecdn.com/ss_thumbnails/codedagentsdeck-251215155422-5497c599-thumbnail.jpg?width=640&height=640&fit=bounds)















