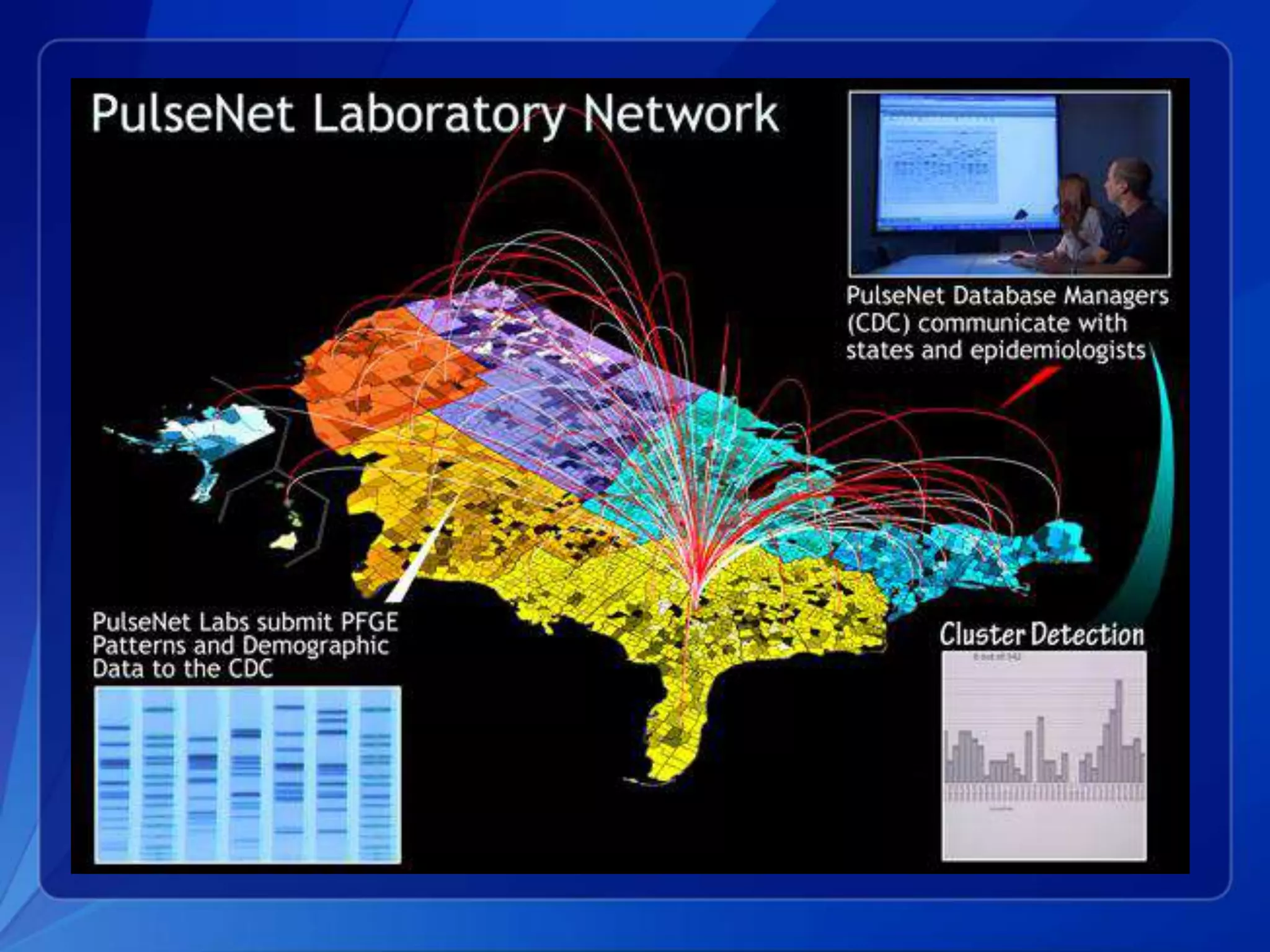
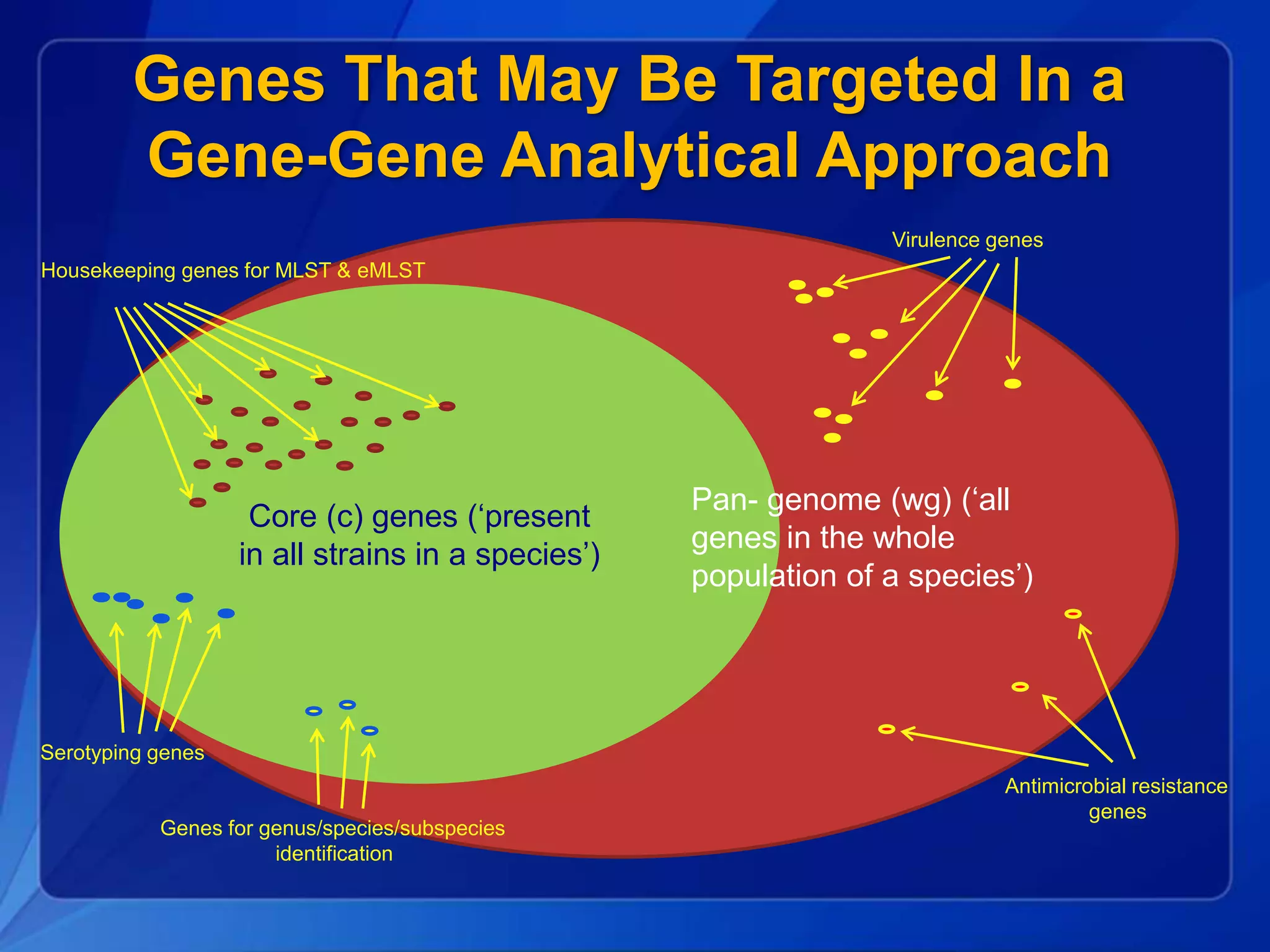
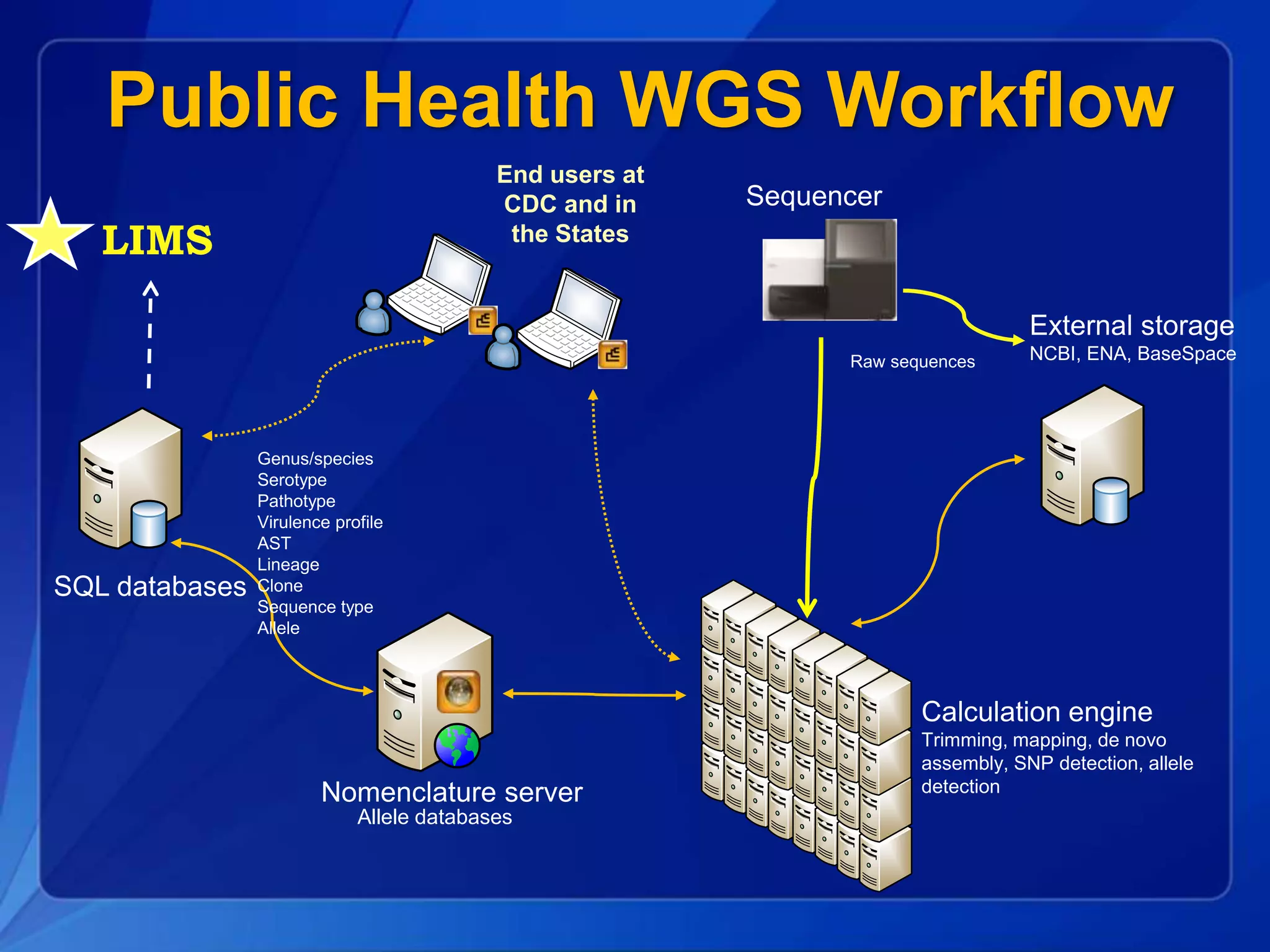
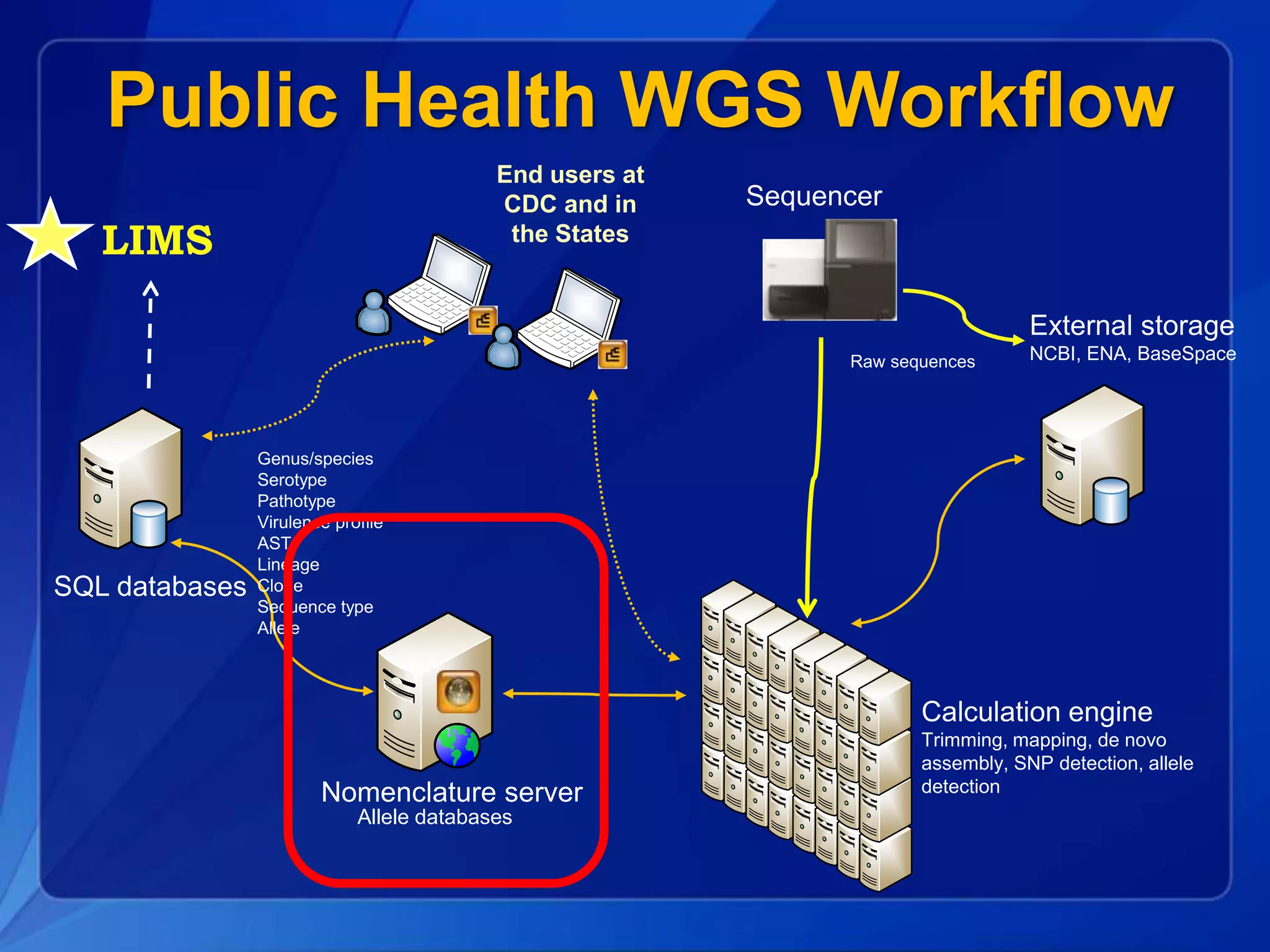
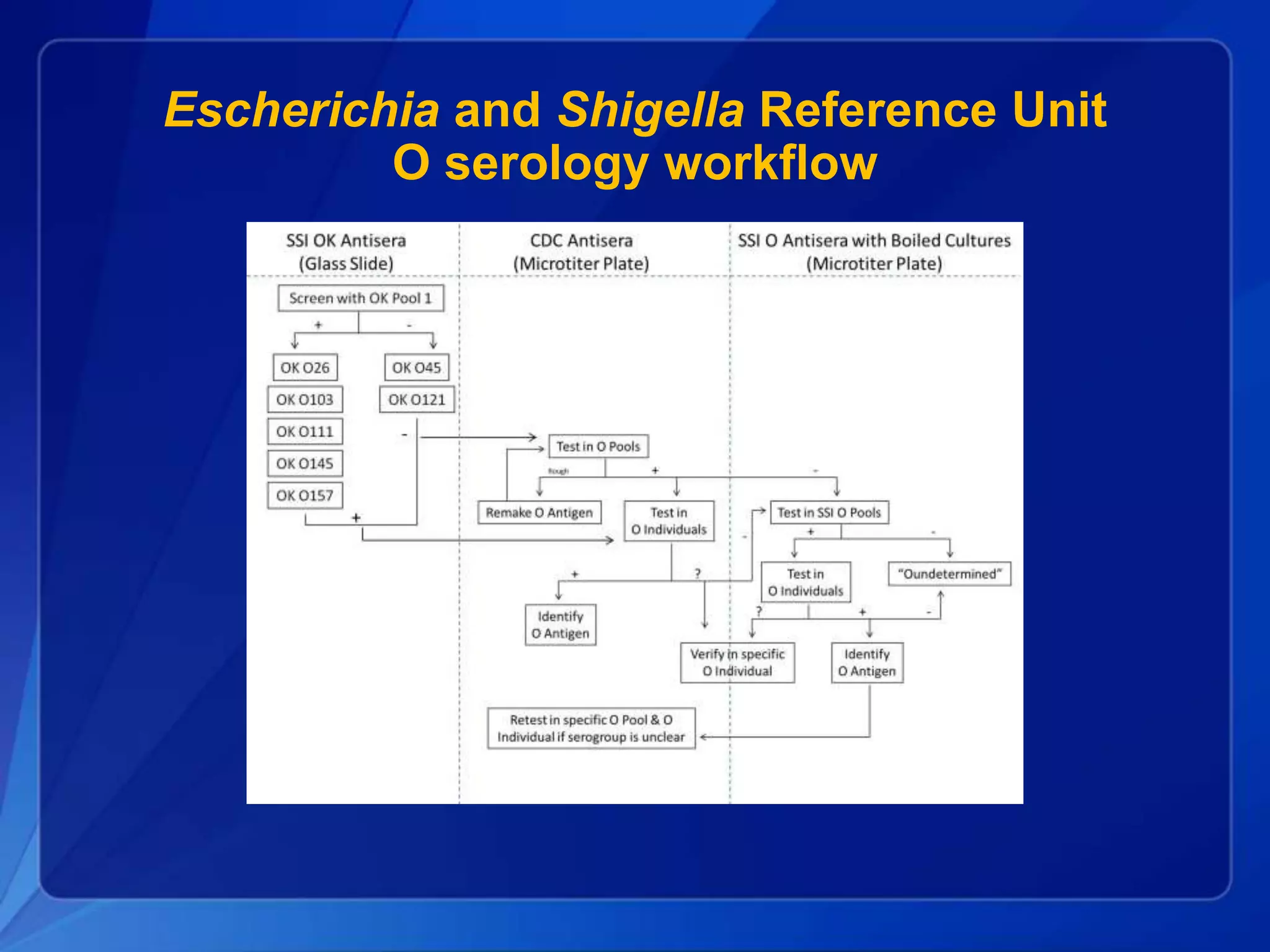
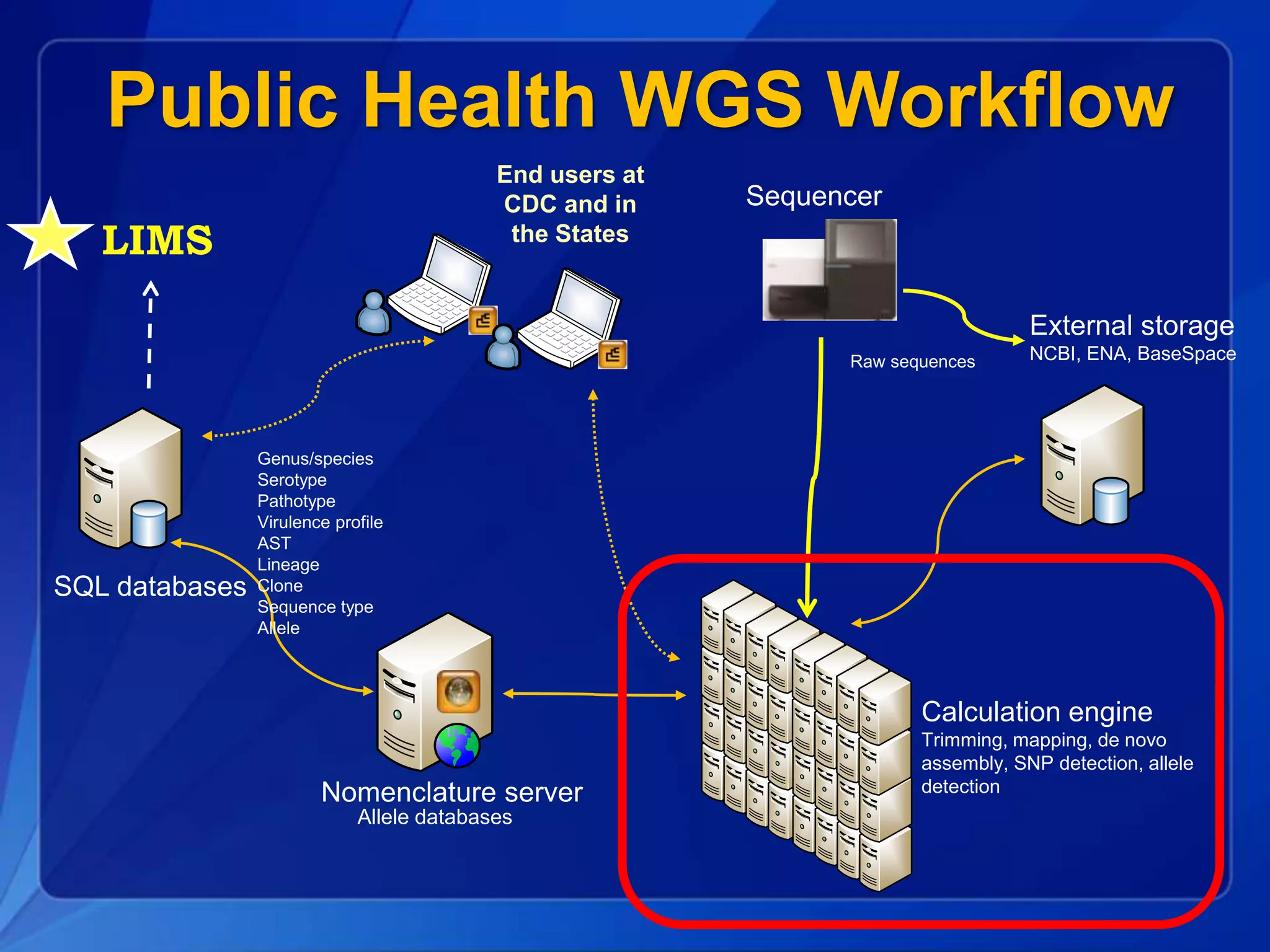
The document discusses the implementation of next-generation sequencing (NGS) for the identification and subtyping of foodborne pathogens, aiming to enhance public health microbiology by consolidating multiple workflows into a single, efficient system. It outlines collaborations among various public health organizations and emphasizes the importance of standardization in methods, nomenclature, and analysis to improve outbreak detection and pathogen characterization. Additionally, the document highlights the need for comprehensive bioinformatics tools and workflows to facilitate real-time data sharing and analysis in public health laboratories.