This document provides an overview and comparison of popular next-generation sequencing platforms. It discusses the common sequencing pipeline including library preparation, massively parallel sequencing, and bioinformatics analysis. Popular platforms like Roche 454, Illumina, and SOLiD are described in detail focusing on their specific sequencing chemistries and performance characteristics. Newer third-generation platforms such as Ion Torrent, PacBio, and Oxford Nanopore are also introduced. A wide range of NGS applications from whole genome sequencing to RNA-seq are outlined.
In this document
Powered by AI
Introduction to NGS and its platforms including Roche, AB SOLiD, Illumina, Ion Torrent, PacBio, and Oxford Nanopore.
Discusses the NGS pipeline from DNA sample to data analysis. Includes library preparation and data flow.
Details the process of library preparation, PCR bias issues, and its impact on quantitative applications.
Describes how NGS combines DNA synthesis with detection and the challenges related to sequencing errors.
Comparison of various sequencing platforms detailing their read lengths, run times, advantages, and disadvantages.
Explains the emulsion PCR process for Roche 454 library preparation before sequencing.
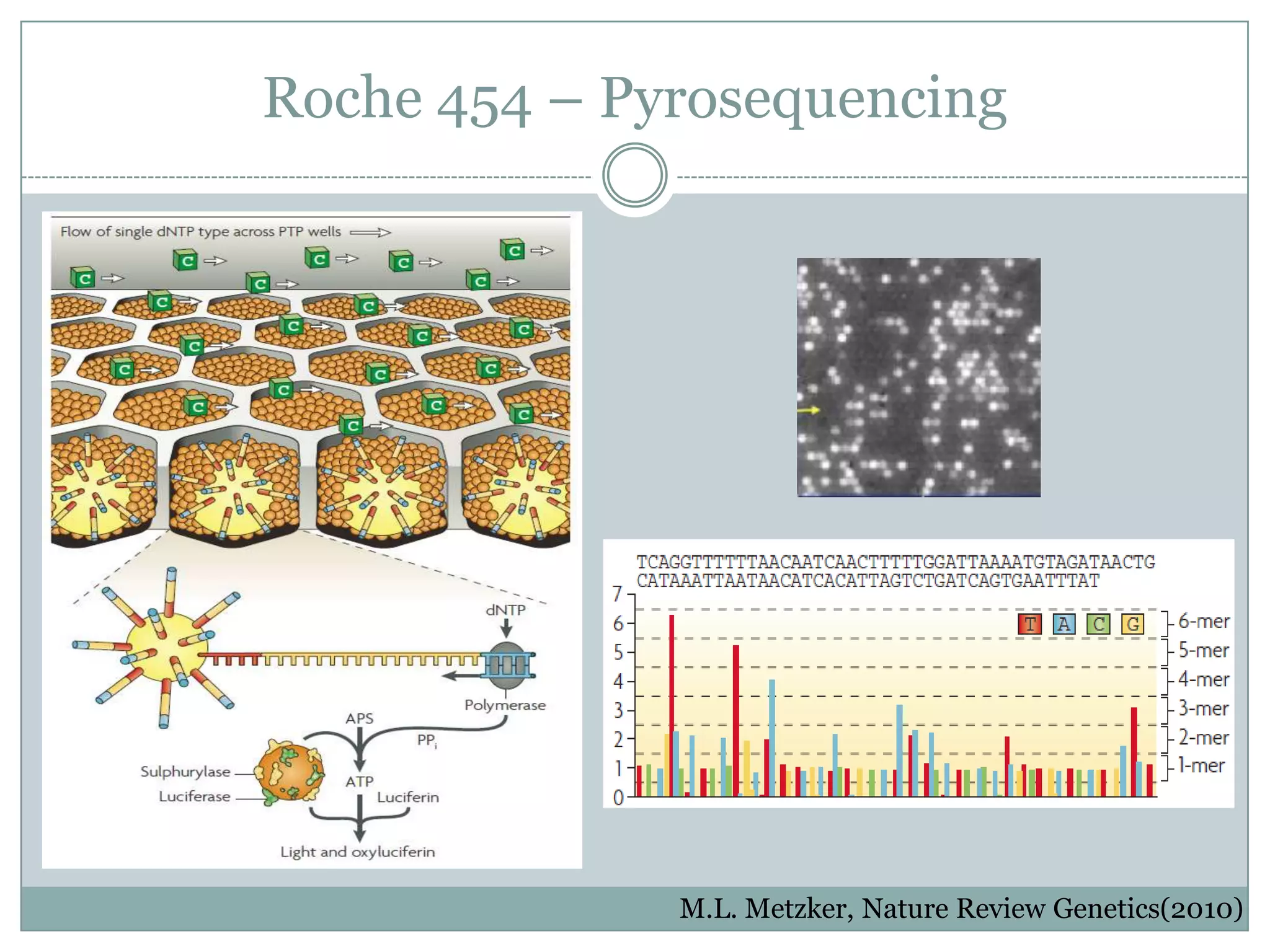
Introduces Roche 454's Pyrosequencing technology and methods used.
Highlights the key steps involved in library preparation for the Illumina sequencing platform.
Details the steps in Illumina's sequencing process, including error rates and yield.
Summarizes the high output and versatility of Illumina, along with its error rates and quality concerns.
Describes SOLiD's library prep, two-base encoding, and ligation detection methods.
Explains SOLiD's approach to color space and its implications for sequencing accuracy.
Discusses SOLiD's low error rates for SNP detection, benefits, and read length limitations.
Describes Ion Torrent's pH detection methodology, advantages, and challenges with homopolymer reads.
Explains PacBio's single molecule sequencing, real-time detection, advantages, and challenges.
Introduces Oxford Nanopore technology and its potential for fast sequencing and long read lengths.
Describes NGS applications including whole genome, exome sequencing, RNA-seq, and SNP discovery.
Concludes the presentation, thanking contributors and listing references.
NGS Overview PartI:
A Comparison of Next-Generation
Sequencing Platforms.
MIN SOO KIM
APRIL 1, 2013
QUANTITATIVE BIOMEDICAL RESEARCH CENTER
DEPARTMENT OF CLINICAL SCIENCES
UT SOUTHWESTERN
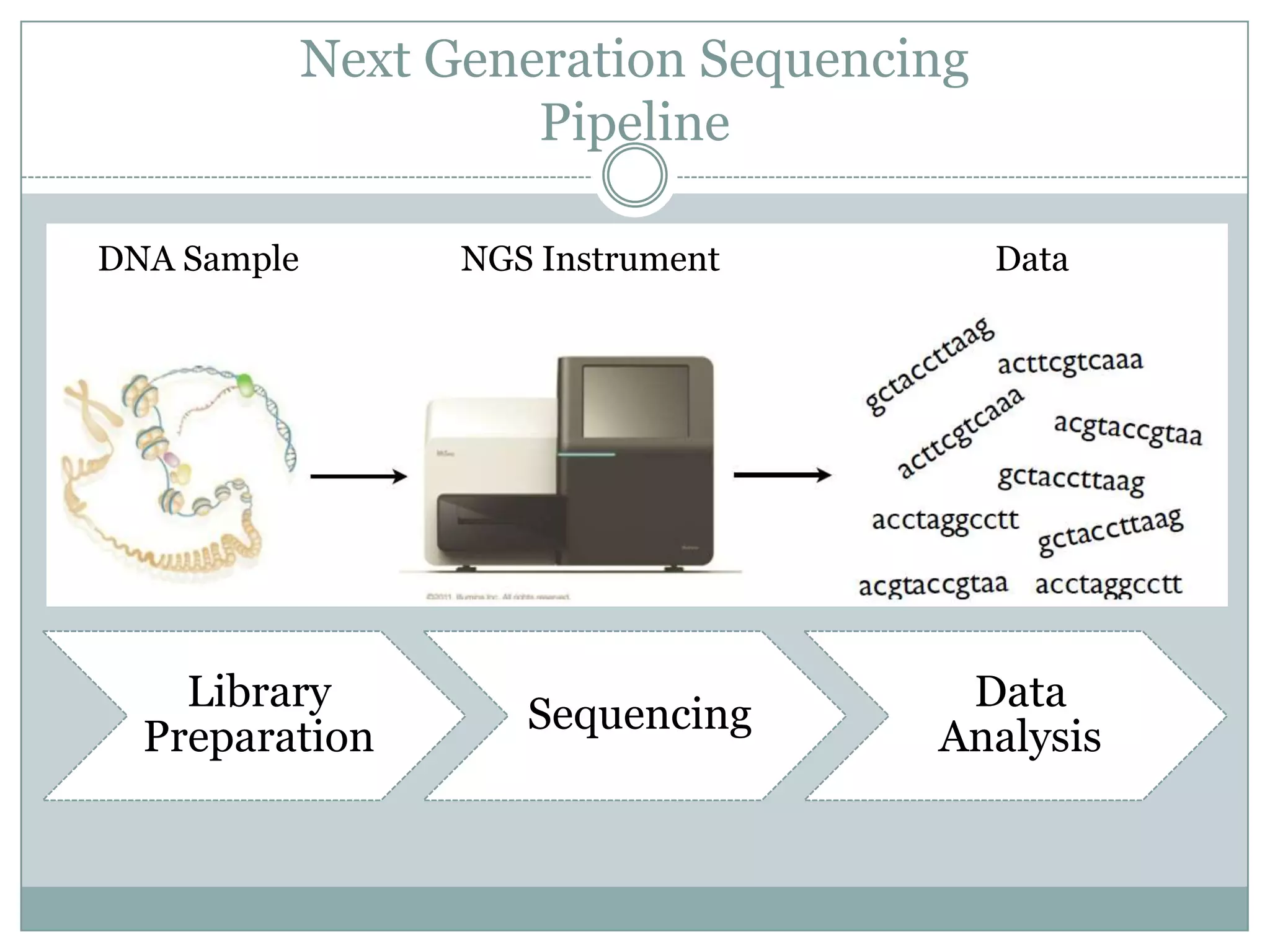
Next Generation Sequencing
Pipeline
DNA Sample NGS Instrument Data
Library Data
Sequencing
Preparation Analysis
5.
Library Preparation
DNAsamples are randomly fragmented and
platform-specific adaptors are added to the flanking
ends to produce a “library”.
Library is then amplified through PCR. (Platform-
specific amplification e.g. beads or glass)
Amplification Introduces Bias:
Amplification bias against AT, GC rich regions. (corrected by
adding PCR additives.)
Alteration of representational abundances(duplicates).
Important for quantitative applications like RNA-seq.
Overrepresentation of smaller fragments. (corrected by running
fewer PCR cycles.)
6.
Massively Parallel Sequencing
In Sanger-sequencing the DNA synthesis and
detection steps are two separate procedures (slow).
Next generation sequencing relies on coupling the
DNA synthesis and detection (sequencing by
synthesis) and multiple sequencing reactions are run
simultaneously (Massively Parallel Sequencing).
For most NGS platforms desynchronization of reads
during the sequencing and detection cycle is the
main cause of sequencing errors and shorter reads.
7.
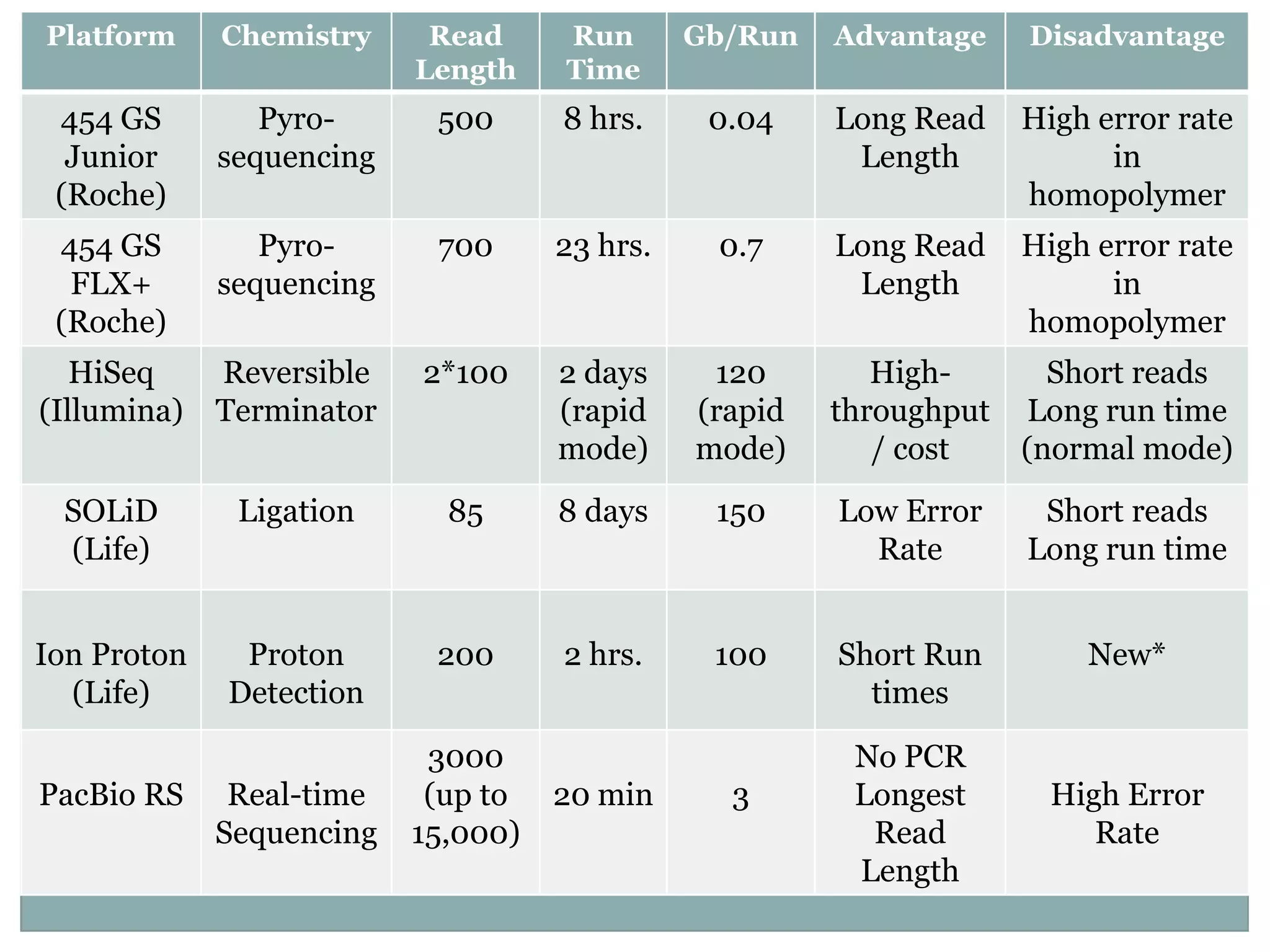
Platform Chemistry Read Run Gb/Run Advantage Disadvantage
Length Time
454 GS Pyro- 500 8 hrs. 0.04 Long Read High error rate
Junior sequencing Length in
(Roche) homopolymer
454 GS Pyro- 700 23 hrs. 0.7 Long Read High error rate
FLX+ sequencing Length in
(Roche) homopolymer
HiSeq Reversible 2*100 2 days 120 High- Short reads
(Illumina) Terminator (rapid (rapid throughput Long run time
mode) mode) / cost (normal mode)
SOLiD Ligation 85 8 days 150 Low Error Short reads
(Life) Rate Long run time
Ion Proton Proton 200 2 hrs. 100 Short Run New*
(Life) Detection times
3000 No PCR
PacBio RS Real-time (up to 20 min 3 Longest High Error
Sequencing 15,000) Read Rate
Length
8.
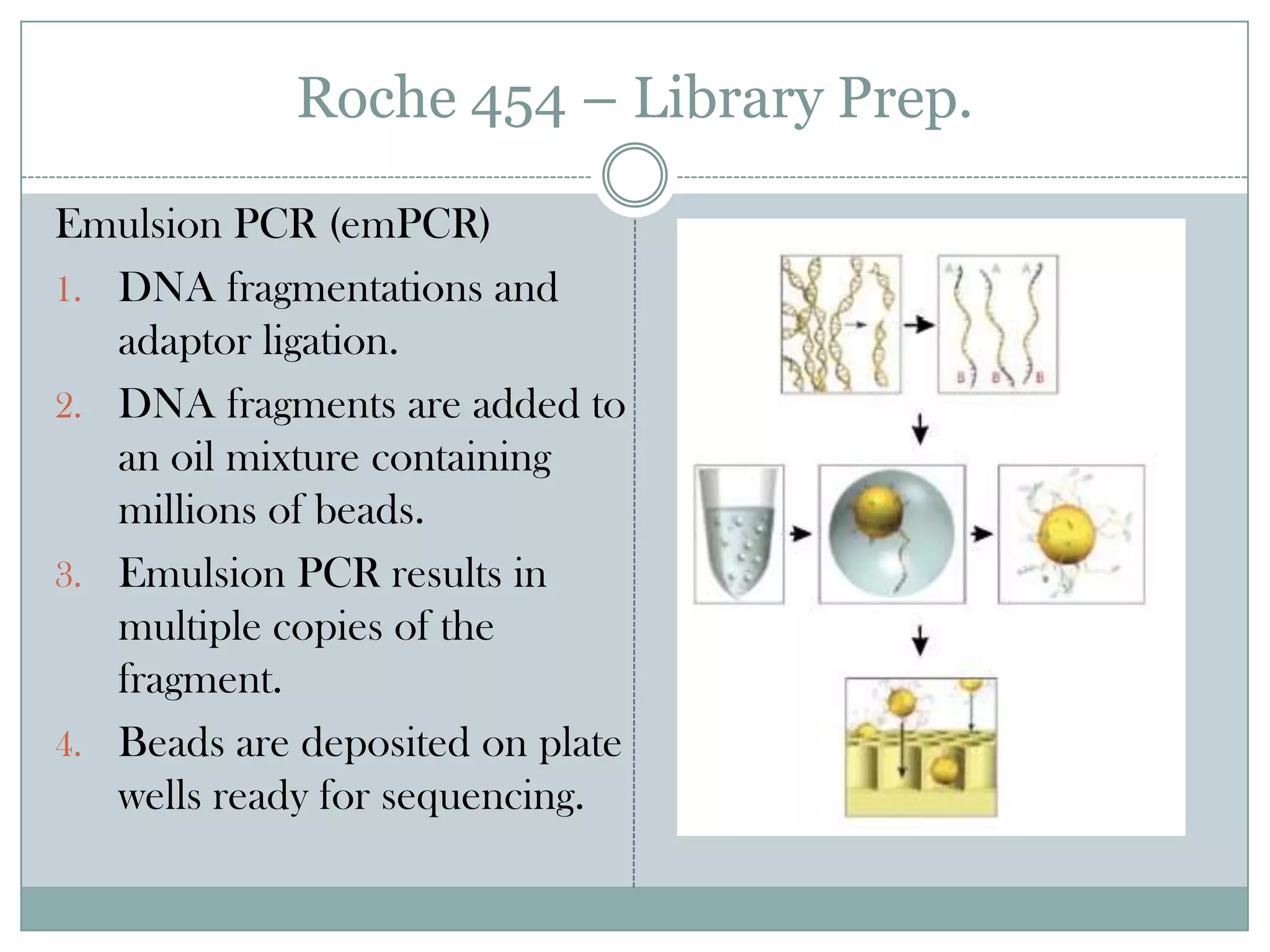
Roche 454 –Library Prep.
Emulsion PCR (emPCR)
1. DNA fragmentations and
adaptor ligation.
2. DNA fragments are added to
an oil mixture containing
millions of beads.
3. Emulsion PCR results in
multiple copies of the
fragment.
4. Beads are deposited on plate
wells ready for sequencing.
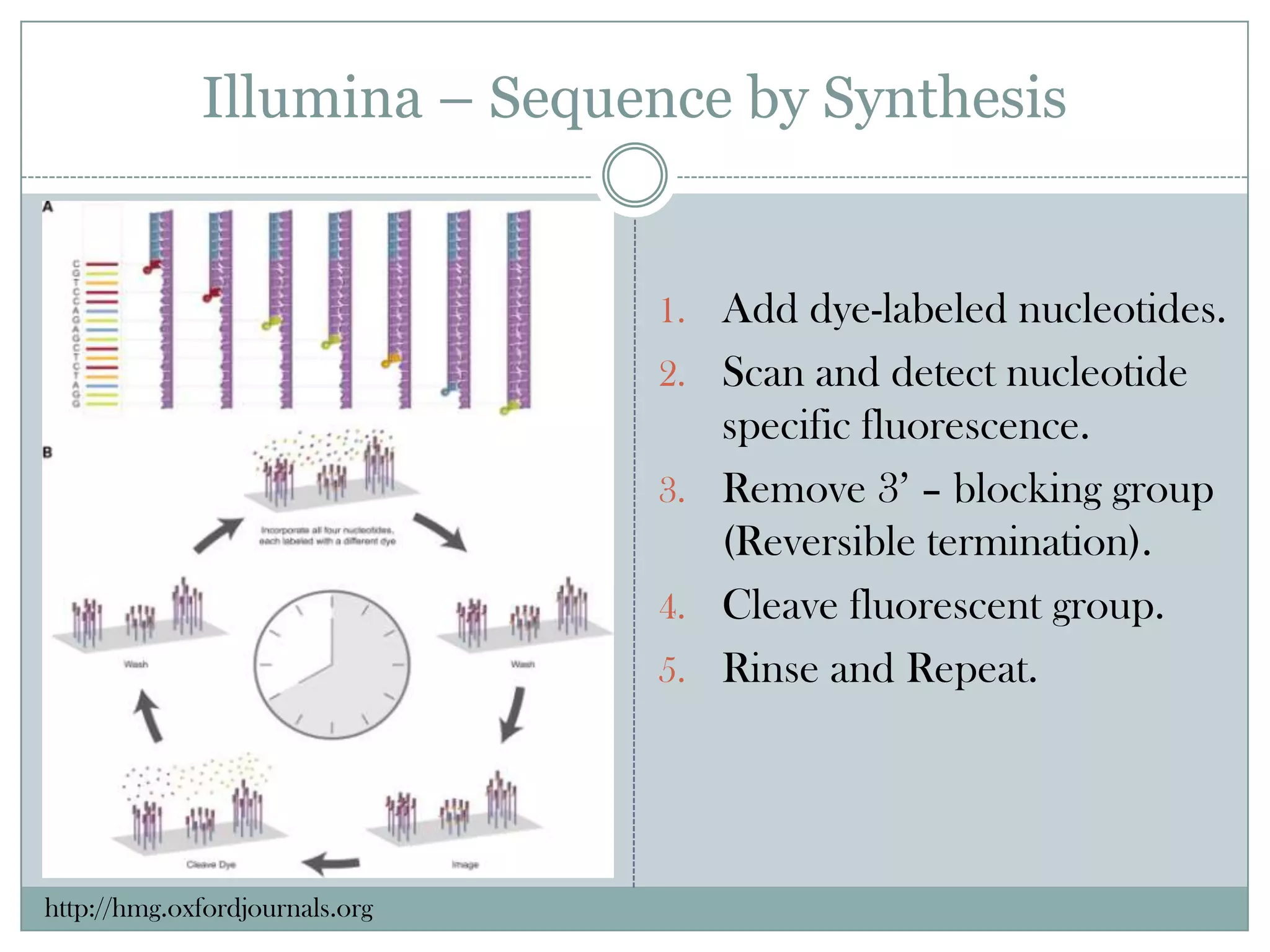
Illumina – Sequenceby Synthesis
1. Add dye-labeled nucleotides.
2. Scan and detect nucleotide
specific fluorescence.
3. Remove 3’ – blocking group
(Reversible termination).
4. Cleave fluorescent group.
5. Rinse and Repeat.
http://hmg.oxfordjournals.org
12.
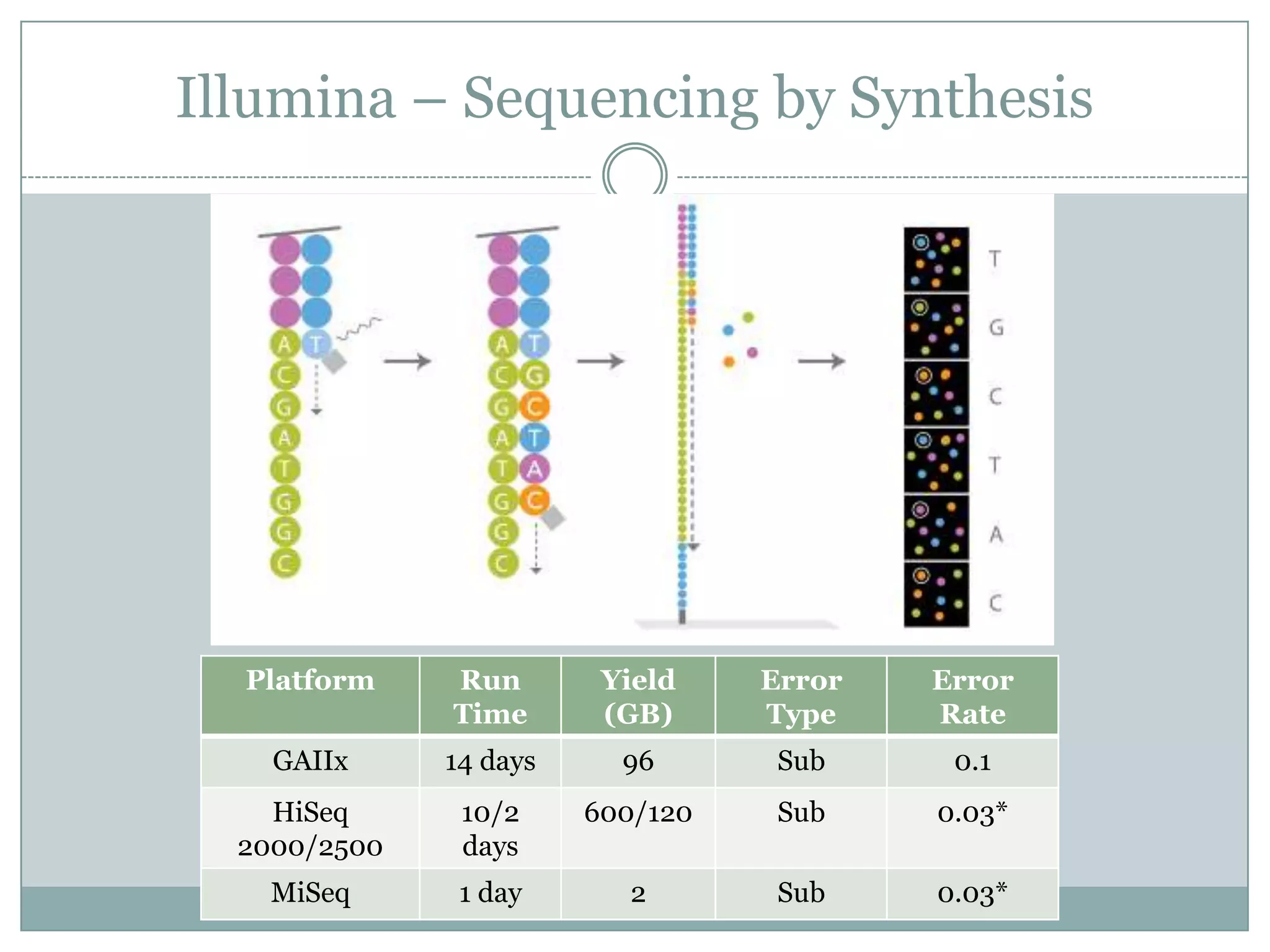
Illumina – Sequencingby Synthesis
Platform Run Yield Error Error
Time (GB) Type Rate
GAIIx 14 days 96 Sub 0.1
HiSeq 10/2 600/120 Sub 0.03*
2000/2500 days
MiSeq 1 day 2 Sub 0.03*
13.
Illumina
Advantages
High throughput/ cost.
Suitable for a wide rage of applications most notably
whole genome sequencing.
Disadvantages
Substitution error rates (recently improved).
Lagging strand dephasing causes sequence quality
deterioration towards the end of read.
14.
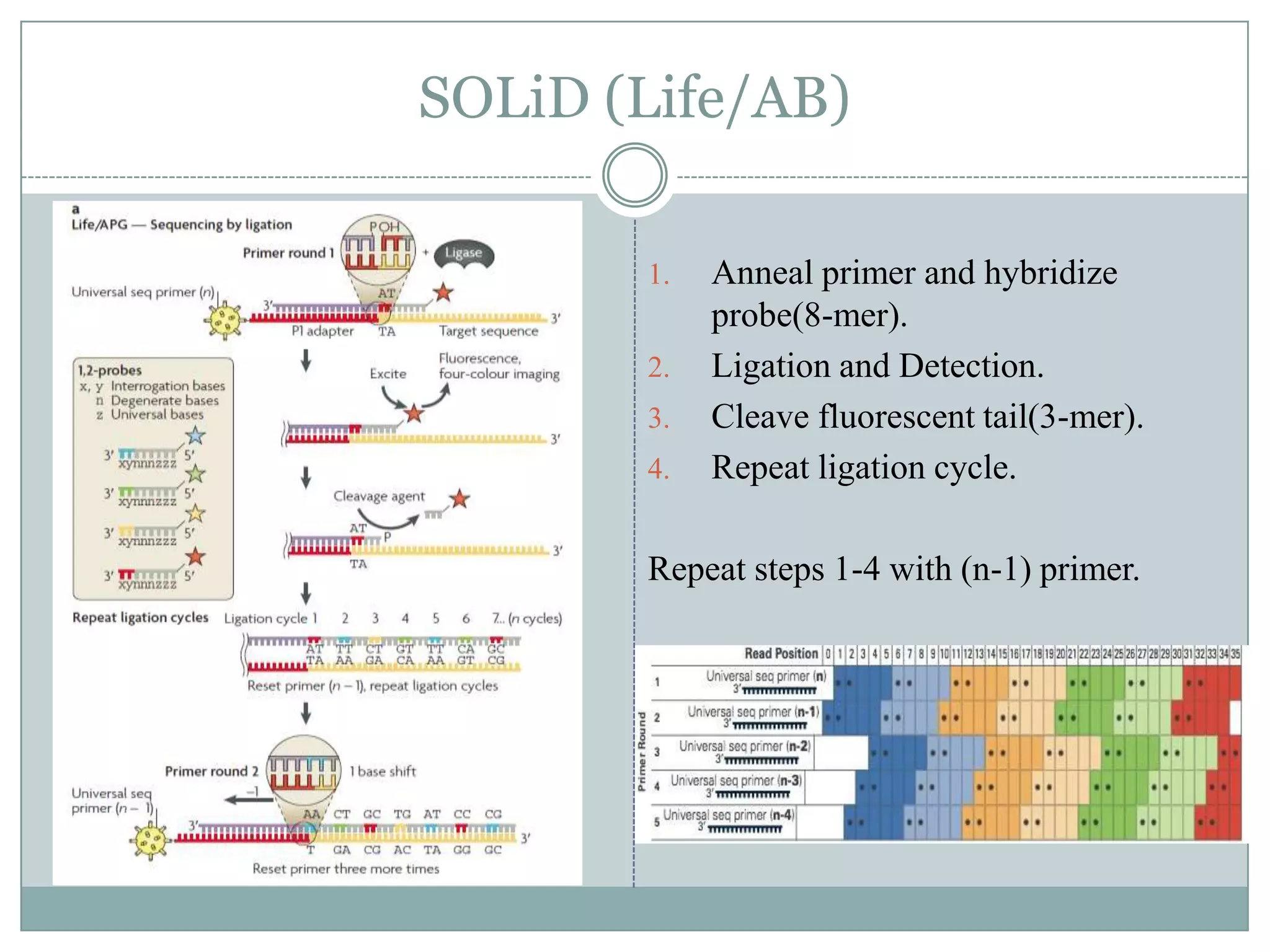
SOLiD (Life/AB)
SOLiD:Sequencing by Oligonucleotide Ligation and
Detection.
Library Prep: emPCR
“2-base encoding” – Instead of the typical single
dNTP addition, two base matching probes are used.
(possible 16 probes).
Color Space – four color sequencing encoding
further increases accuracy.
15.
SOLiD (Life/AB)
1. Anneal primer and hybridize
probe(8-mer).
2. Ligation and Detection.
3. Cleave fluorescent tail(3-mer).
4. Repeat ligation cycle.
Repeat steps 1-4 with (n-1) primer.
16.
SOLiD – ColorSpace
• 16 possible base combination
are represented by 4 colors.
• All possible sequencing
combination need to be decoded.
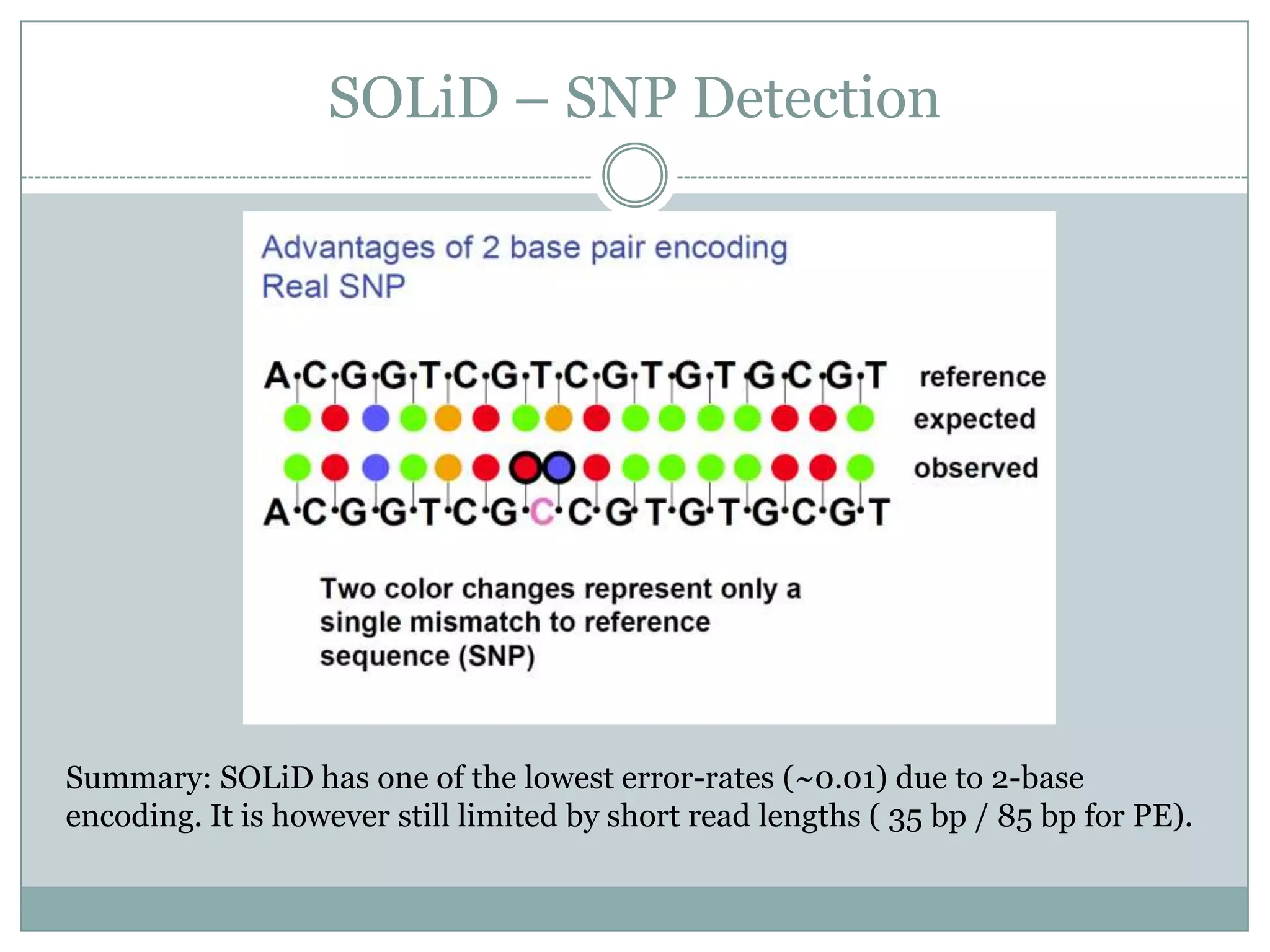
SOLiD – SNPDetection
Summary: SOLiD has one of the lowest error-rates (~0.01) due to 2-base
encoding. It is however still limited by short read lengths ( 35 bp / 85 bp for PE).
19.
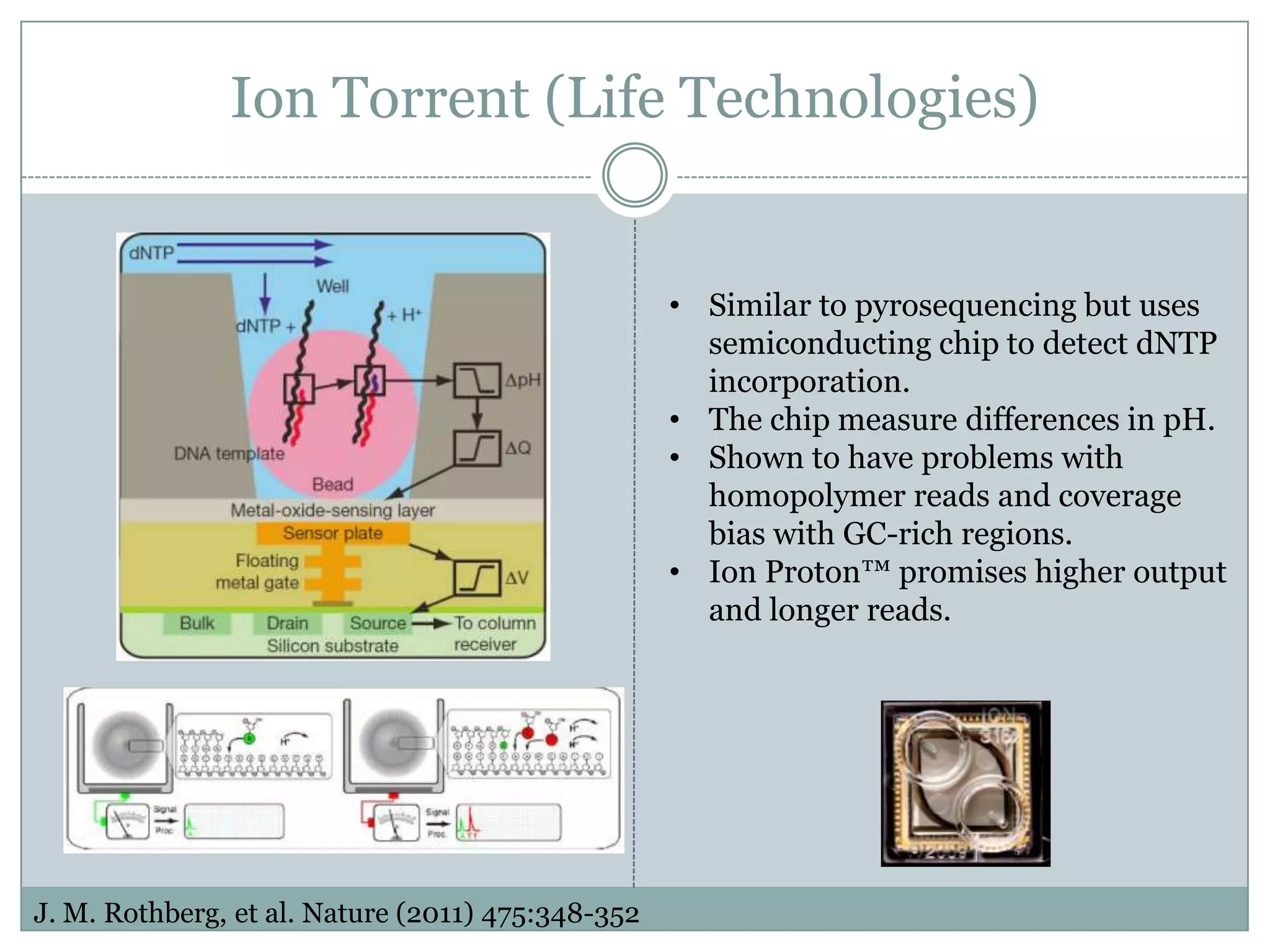
Ion Torrent (LifeTechnologies)
• Similar to pyrosequencing but uses
semiconducting chip to detect dNTP
incorporation.
• The chip measure differences in pH.
• Shown to have problems with
homopolymer reads and coverage
bias with GC-rich regions.
• Ion Proton™ promises higher output
and longer reads.
J. M. Rothberg, et al. Nature (2011) 475:348-352
20.
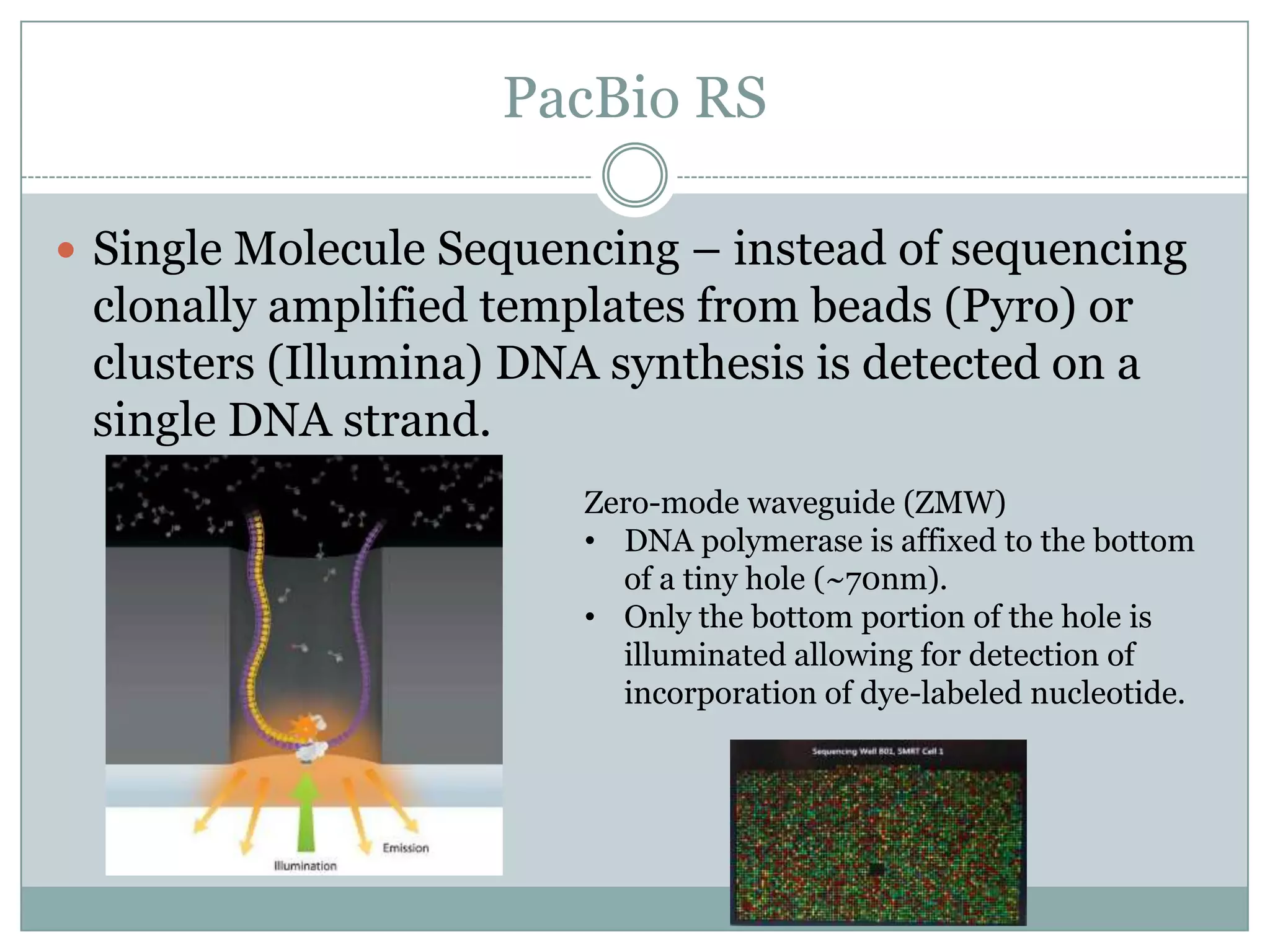
PacBio RS
SingleMolecule Sequencing – instead of sequencing
clonally amplified templates from beads (Pyro) or
clusters (Illumina) DNA synthesis is detected on a
single DNA strand.
Zero-mode waveguide (ZMW)
• DNA polymerase is affixed to the bottom
of a tiny hole (~70nm).
• Only the bottom portion of the hole is
illuminated allowing for detection of
incorporation of dye-labeled nucleotide.
21.
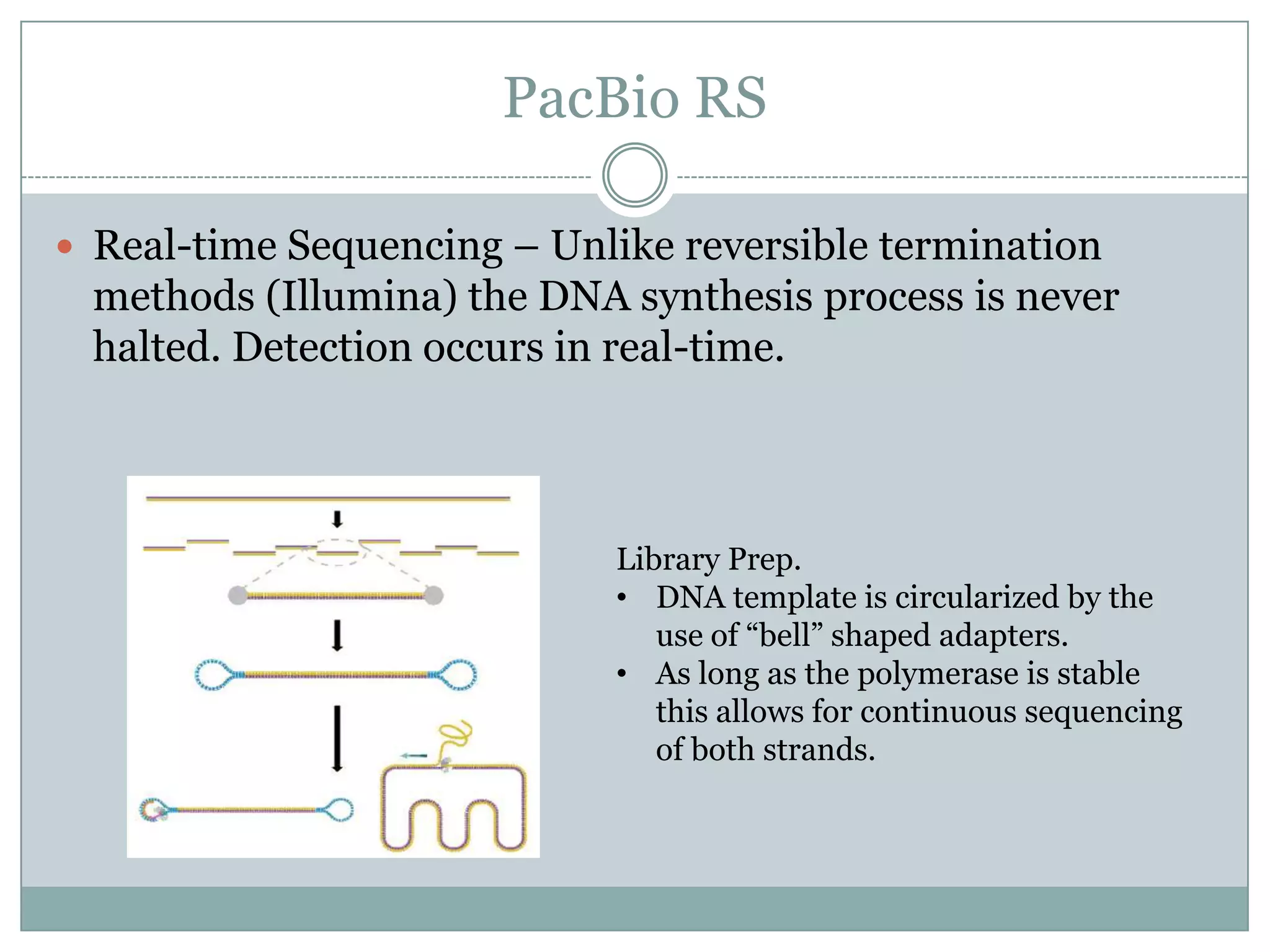
PacBio RS
Real-timeSequencing – Unlike reversible termination
methods (Illumina) the DNA synthesis process is never
halted. Detection occurs in real-time.
Library Prep.
• DNA template is circularized by the
use of “bell” shaped adapters.
• As long as the polymerase is stable
this allows for continuous sequencing
of both strands.
22.
PacBio RS
Advantages
Noamplification required.
Extremely long read lengths.
Average 2500 bp. Longest 15,000bp.
Disadvantages
High error rates.
Error rate of ~15% for Indels. 1% Substitutions.
23.
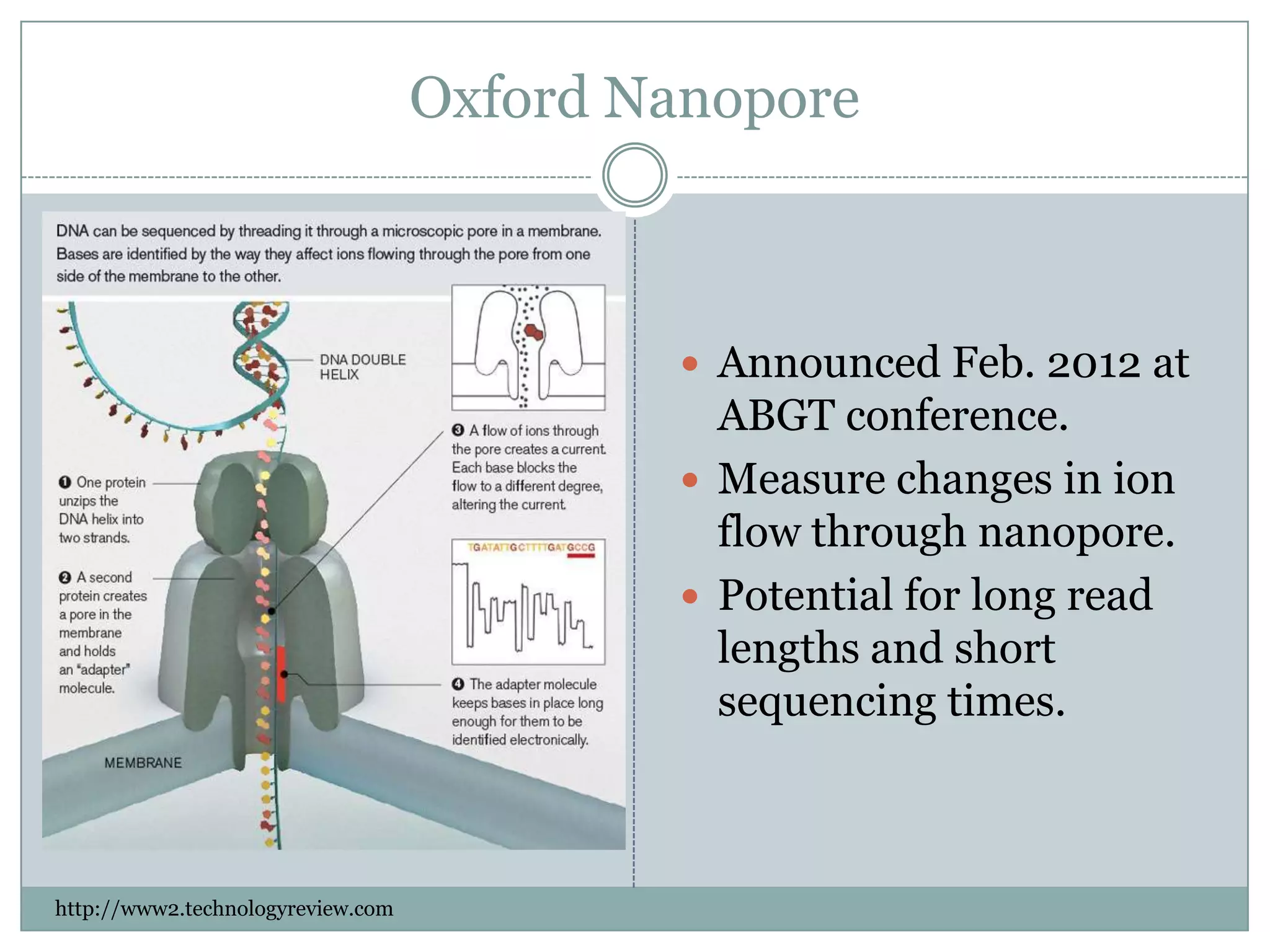
Oxford Nanopore
Announced Feb. 2012 at
ABGT conference.
Measure changes in ion
flow through nanopore.
Potential for long read
lengths and short
sequencing times.
http://www2.technologyreview.com
Thank you.
Acknowledgements
Dr. TaeHyun Hwang
References
Mardis, E. R. A decade’s perspective on DNA sequencing technology.
Nature (2011) 470:198 - 203
Metzker, M.L. Sequencing technologies – the next generation. Nature
Review Genetics(2010) 11:31 – 46
N. J. Loman, C. Constantinidou, Jacqueline Z. M. Chan, M. Halachev, M.
Sergeant, C. W. Penn, E. R. Robinson & M. J. Pallen. High-throughput
bacterial genome sequencing: an embarrassment of choice, a world of
opportunity. Nature Review Microbiology 10, 599-606.
Editor's Notes
#2 Thank you for joining us in our tech talk series. Today’s tech talk will be an overview of next generation sequencing platforms.
#3 I will start of by giving a brief history on the origin of next generation sequencing. I will then cover some of the common steps in doing next-gen sequening. We will then cover in more detail the specific technologies behind some of the more popular sequencing platforms we use as well discuss some of the new comers in the sequencing Market. These next generation sequencing technologies have been mostly developed and sold by individual companies. I am however not affiliated with any of them. I will try to present both the advantages and disadvantage of each system.
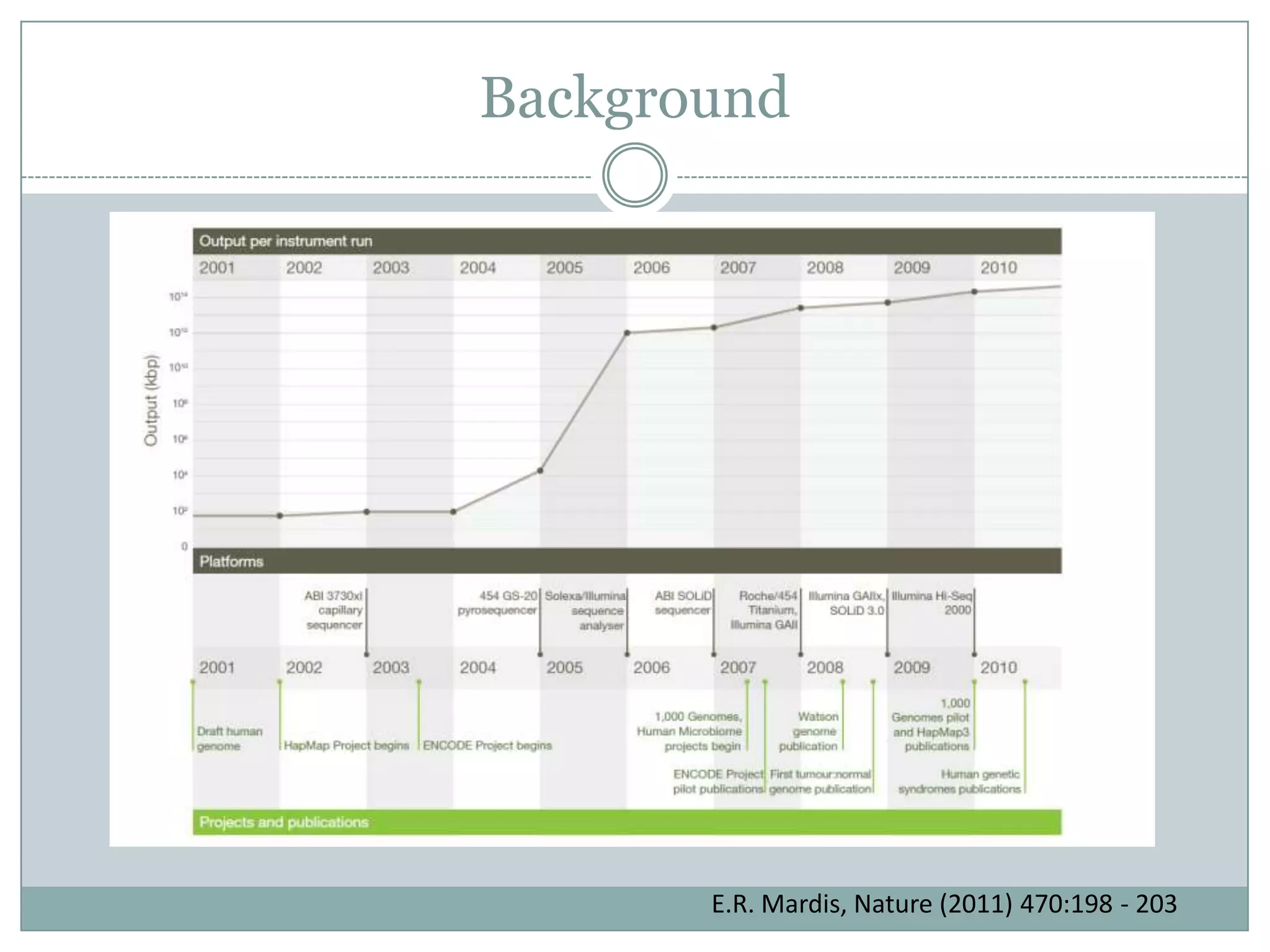
#4 Although it has become quite common to do nex-gen sequencing studies many biomedical research labs, the technologies behind the data have really only come about in the past decade. The story of next generation sequencing start with the Human Genome Project.2000 First draft of Human Genome Complete. - This set the roadmap for modern biomedical research. - Most of the DNA sequencing for the Human Genome Project was based on Sanger Sequencing. - Sanger Sequencing methods were slow and expensive. Estimated cost of the human genome project is 3 billion dollars and 10 years to generate the first draft of Human Genome. - After the completion of the first Human Genome there was an increased focus on improving the machinery used to generate sequencing data.Introduction of Next Generation Sequencing Instruments. - Next Generation Sequencing refers to the advancement of sequencing technologies that allowed for significant improvements in quantity, speed, and cost of generating sequencing data. - In 2004 first parallelized version of 454 pyrosequencing instrument was commercialize. 6-fold cost reduction compared to capillary based Sanger Sequencing. - Since then a wide range of similar sequencing technologies have been introduced bringing down the cost even further. - We are now getting very close to the $1000 per sample mark for Whole Genome Sequencing.
#6 Single-molecule template sequencing avoids some of these problems by circumventing the amplification step all together. I will go over an example of a platfDNA amplification results in multiple copies of the initial DNA fragment. For most NGS platforms, except for single molecule sequencing instruments, this is necessary because sequencing takes places on all the fragments at the same time. The sequencing signal from all the fragments are read in at once. This is also the main reason why the read lengths of most NGS platforms are short. Incomplete extension or over extension of the sequencing stand can result in strand dephasing which causes noise in the detection signal. Accumulation of dephased strands deceases the signal to noise ratio to a point where the sequence can no longer be read causing the termination of the sequencing read. Real-time sequencers can avoid this problem leading to much longer sequencing reads, but they suffer from other limitations ( which I will go over later. ) ormthat uses single-molecule sequencing.
#7 The main purpose for dna amplification is so that you can sequence all the fragments at once. This is essentially what sets Next generation sequencing apart from Sanger. In Sanger …
#8 SOLiD has lowest error rate. Good for resequencing.Illumina has the highest throughput / cost.PacBio has the longest read length. Up to 15,000 bp. Applications in DeNovo sequencing and structural variation studies.Ion Torrent from Life Technologies has two instruments. The Personal Genome Machine which has been out since 2010. It’s mostly only applicable for small genome sequencing like bacterial genomes. The Ion Proton, which promises much greater throughput and it has been recently made commercially available. There is however limited information on it’s actual performance.
#9 Emulsion PCR Fragment DNA and attache adapters.PCR is carried out in a oil mixture containing beads and reagents.Each of the beads has a small adaptor that is complementary to the adaptor attached to the DNA fragment.Once the DNA is added to the emulsion ideally you get one fragment attached to each bead.You then carry out the PCR reaction which results in multiple clonal DNA fragments on each bead.The beads are then lowered on plates containing millions of wells roughly the size of each bead.
#10 Additional beads are added to each well containing Sulphurulase and Luciferase.Reagents are flowed through the top of the plates for the sequencing reaction to occur. A key step to the process is that only one nucleotide type (ex: cytosine) is added to the plate.If the added nucleotide is complementary to the template strand it is incorporated into the growing strand by the polymerase releasing a pyrophosphate.The pyrophosphate is then picked up by the catalytic enzymes causing a chain reaction which ultamately emits light.The light is then picked up by CCD camera and recoded on a flowgram.The cycle repeated between reagent and washing.A key limitation to this procedure is that if you have a chain of identical nucleotides on the template sequences you will get the addition of multiple nucleotides in a single reaction cycle. The light intensity will be proportional to the number of nucleotides added but the detection limit is around 6 or 7. The camera will therefore not be able to detect homopolymer runs that are longer than 6 or 7 nucleotides.The most common error rate in pyrosequencing is therefore indels which is around 1%. Substitution error rate however are very low which makes this platform great for targeted resequencing and studies where you are looking for single nucleotide substitutions.
#11 Illumina Library Prep is done through a process called bridge amplification.1. PCR is done on glass plate called flow cells.2. DNA is fragmented and ligate adaptors are added to both ends similar to emulsion PCR.3. Single stranded fragments are attached to surface of flowcells. The flowcell contains the complementary primers to the fragment adaptors.4. PCR reaction is carried out which extents the bridge.5. Through the PCR cycle multiple single strand copies are made creating a dense cluster on the flowcell.
#13 Improve chemistry and detection for Illumina HiSeq/MiSeq
#19 Summary – Due to the 2-base encoding system you get very low error rate. (Error rate of around .01)It is still however limited by short reads. (35bp)
#21 Obvious advantage is no need for clonal amplification. Reduces amplification bias.
#23 Indels errors occur when multiple nucleotide are incorporated too fast for the camera to detect.Improvments to the technology primarily have come with advances in it’s chemistry. Primarily it’s polymerase enzyme. Improvements include higher nucleotide incorporation accuracy. Higher tolerance to heat ( longer reads ).