Downloaded 1,084 times






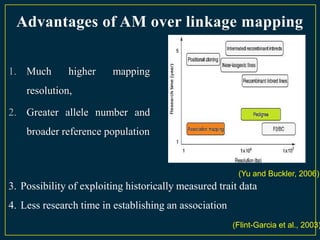






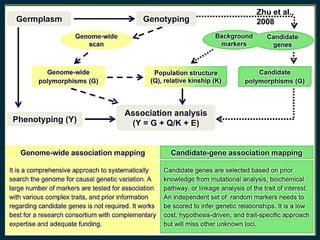
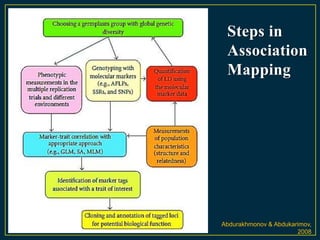










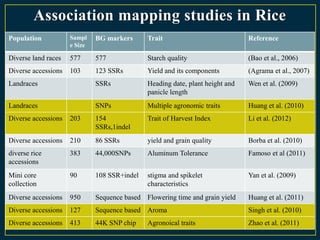

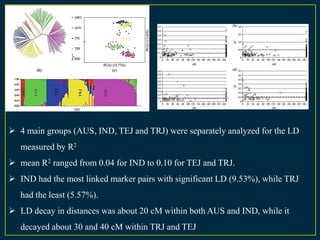
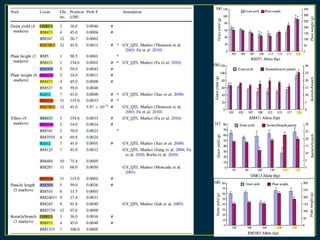

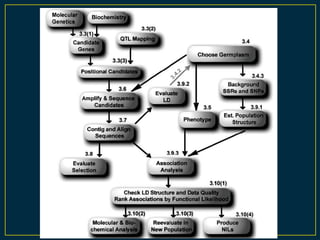









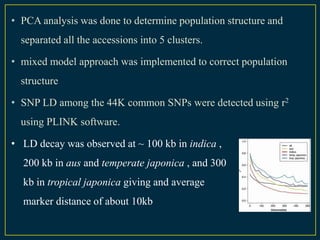




The document discusses advanced methods in crop improvement, focusing on the genetic basis of economically important traits and techniques for mapping quantitative trait loci (QTLs). It compares association mapping (AM) and linkage mapping, highlighting AM's advantages such as higher resolution and broader allele diversity. The text also describes specific software tools and approaches used for detecting associations between genotypes and phenotypes in various plant species.











































































































