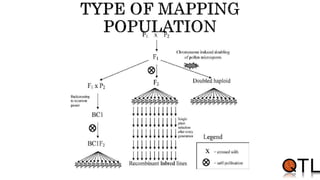
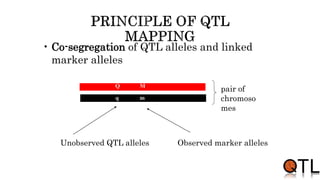
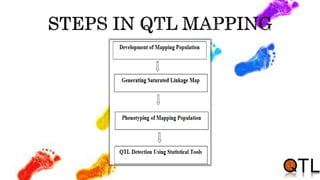
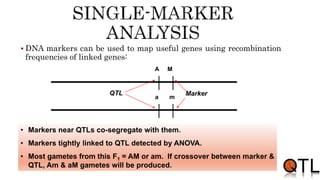
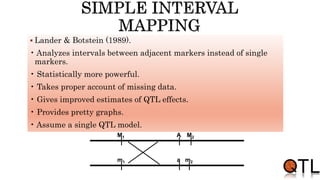
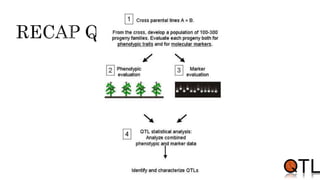
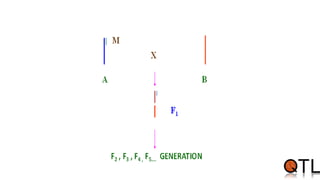This document discusses quantitative trait loci (QTL) mapping. It begins by defining QTLs as genomic regions containing genes associated with quantitative traits. QTL mapping involves correlating genotypic and phenotypic data from a mapping population to identify these regions. Common mapping populations discussed include recombinant inbred lines, double haploids, and backcrosses. Interval mapping and composite interval mapping are presented as methods for QTL analysis. The goals of QTL mapping are to locate genomic regions influencing traits and estimate the effects of QTLs.