Downloaded 14 times























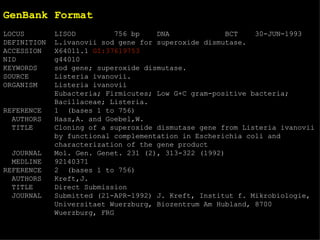

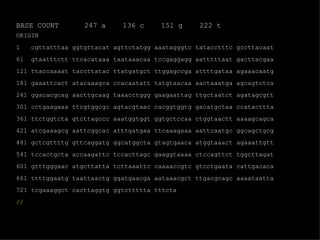
![EMBL format ID LISOD standard; DNA; PRO; 756 BP. IDentification XX AC X64011; S78972; Accession (Axxxxx, Afxxxxxx), GUID XX NI g44010 Nucleotide Identifier --> x.x XX DT 28-APR-1992 (Rel. 31, Created) DaTe DT 30-JUN-1993 (Rel. 36, Last updated, Version 6) XX DE L.ivanovii sod gene for superoxide dismutase DEscription XX. KW sod gene; superoxide dismutase. KeyWord XX OS Listeria ivanovii Organism Species OC Eubacteria; Firmicutes; Low G+C gram-positive bacteria; Bacillaceae; OC Listeria. Organism Classification XX RN [1] RA Haas A., Goebel W.; Reference RT "Cloning of a superoxide dismutase gene from Listeria ivanovii by RT functional complementation in Escherichia coli and RT characterization of the gene product."; RL Mol. Gen. Genet. 231:313-322(1992). XX](https://image.slidesharecdn.com/20120301bioinformaticsiiles1-120301003825-phpapp02/85/2012-03-01_bioinformatics_ii_les1-31-320.jpg)
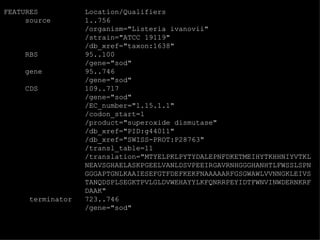
![Example of a SwissProt entry ID TNFA_HUMAN STANDARD; PRT; 233 AA. IDentification AC P01375; ACcession DT 21-JUL-1986 (REL. 01, CREATED) DaTe DT 21-JUL-1986 (REL. 01, LAST SEQUENCE UPDATE) DT 15-JUL-1998 (REL. 36, LAST ANNOTATION UPDATE) DE TUMOR NECROSIS FACTOR PRECURSOR (TNF-ALPHA) (CACHECTIN). GN TNFA. Gene name OS HOMO SAPIENS (HUMAN). Organism Species OC EUKARYOTA; METAZOA; CHORDATA; VERTEBRATA; TETRAPODA; MAMMALIA; OC EUTHERIA; PRIMATES. Organism Classification RN [1] Reference RP SEQUENCE FROM N.A. RX MEDLINE; 87217060. RA NEDOSPASOV S.A., SHAKHOV A.N., TURETSKAYA R.L., METT V.A., RA AZIZOV M.M., GEORGIEV G.P., KOROBKO V.G., DOBRYNIN V.N., RA FILIPPOV S.A., BYSTROV N.S., BOLDYREVA E.F., CHUVPILO S.A., RA CHUMAKOV A.M., SHINGAROVA L.N., OVCHINNIKOV Y.A.; RL COLD SPRING HARB. SYMP. QUANT. BIOL. 51:611-624(1986). RN [2] RP SEQUENCE FROM N.A. RX MEDLINE; 85086244. RA PENNICA D., NEDWIN G.E., HAYFLICK J.S., SEEBURG P.H., DERYNCK R., RA PALLADINO M.A., KOHR W.J., AGGARWAL B.B., GOEDDEL D.V.; RL NATURE 312:724-729(1984). ...](https://image.slidesharecdn.com/20120301bioinformaticsiiles1-120301003825-phpapp02/85/2012-03-01_bioinformatics_ii_les1-32-320.jpg)


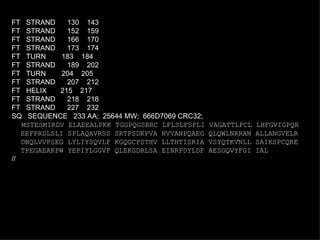










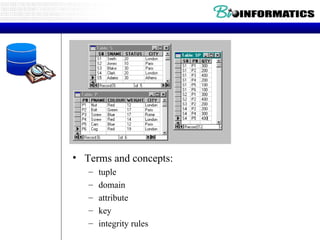



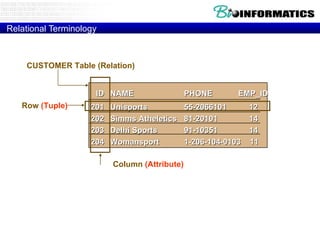

![Relational Database Terminology Relational operators Relational select rel WHERE boolean-xpr project rel [ attr-specs ] join rel JOIN rel divide by rel DIVIDEBY rel Set-based rel UNION rel rel INTERSECT rel \ rel MINUS rel rel TIMES rel](https://image.slidesharecdn.com/20120301bioinformaticsiiles1-120301003825-phpapp02/85/2012-03-01_bioinformatics_ii_les1-52-320.jpg)


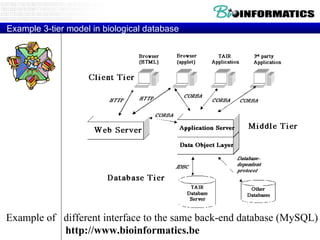










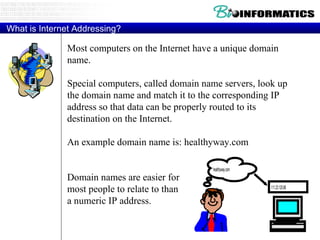




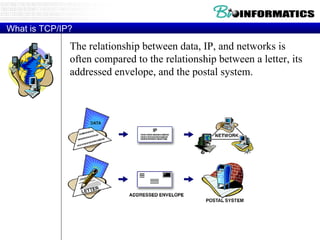








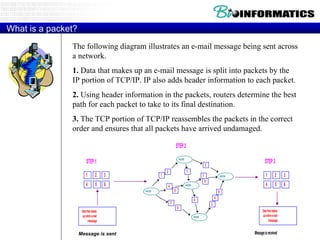

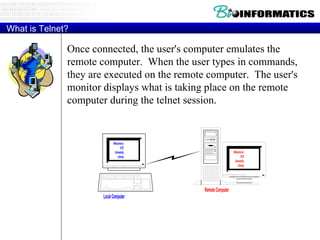















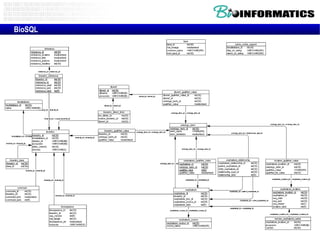


The document provides information about a bioinformatics course including the syllabus, topics, and schedule. The course focuses on using relational databases to manage and analyze large biological datasets. Topics include relation databases, web application development, genome browsers, text mining, and systems biology. The schedule lists the topics to be covered each class over 12 weeks from March to May.

































































![Bio ontologies and semantic technologies[2]](https://cdn.slidesharecdn.com/ss_thumbnails/bioontologiesandsemantictechnologies2-180509123734-thumbnail.jpg?width=640&height=640&fit=bounds)






























