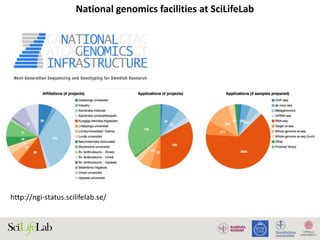
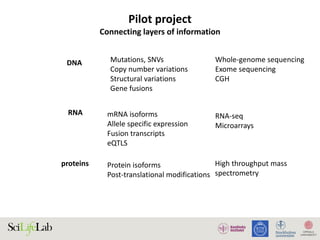
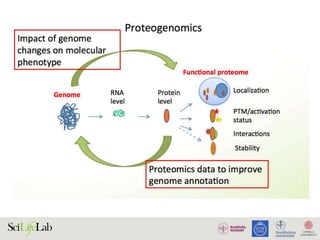
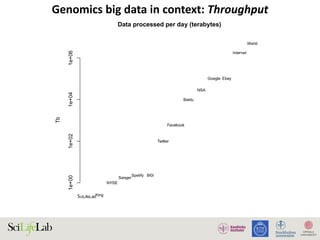
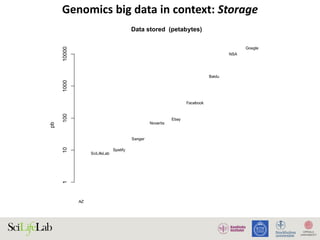
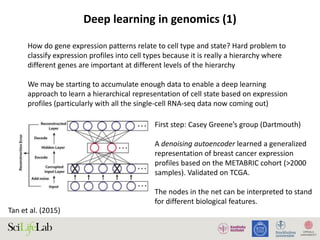
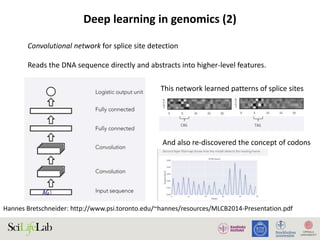
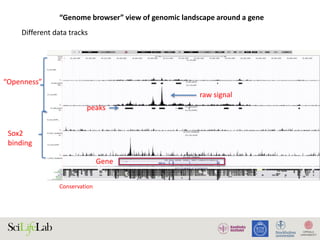
The document discusses data analysis and integration challenges in genomics, focusing on the infrastructure at SciLifeLab in Uppsala and its various research projects. It highlights the importance of bioinformatics, machine learning, and APIs in improving data accessibility and integration for researchers. Additionally, it addresses the significance of data provenance and the limitations of current genomic databases, as well as advances in deep learning that could enhance data interpretation in genomics.