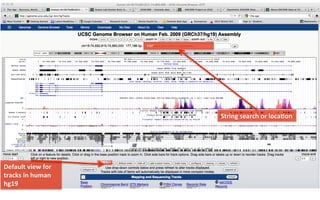
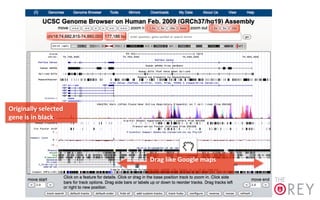
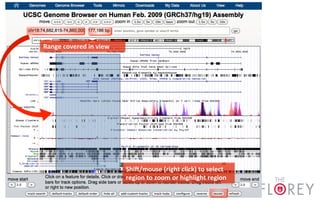
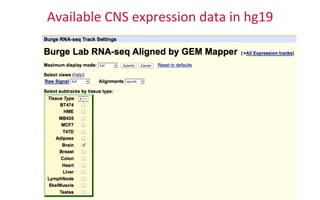
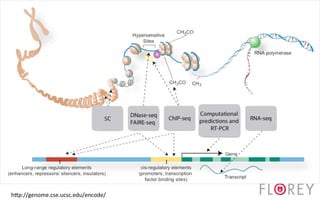
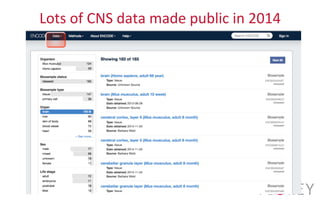
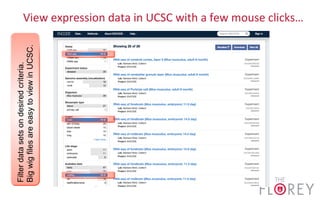
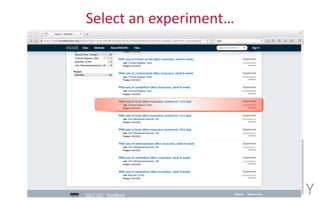
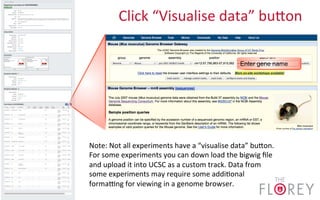
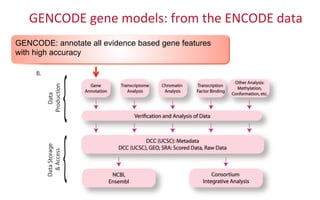
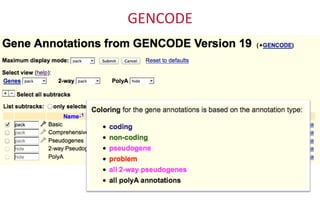
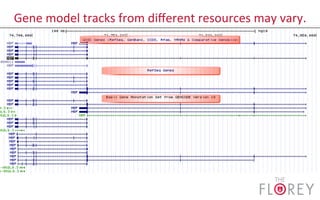
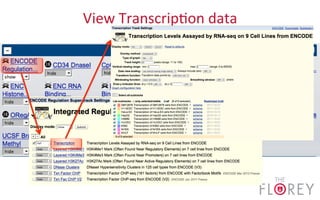
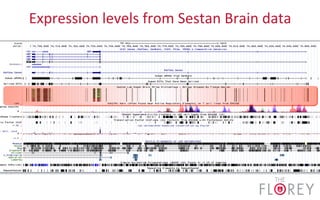
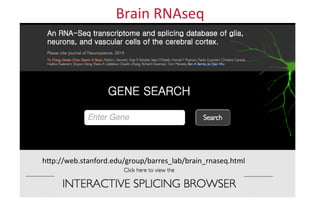
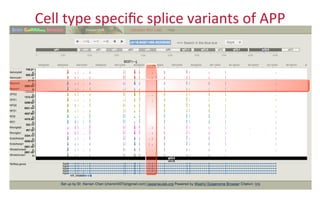
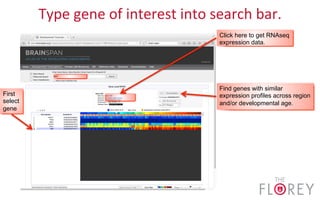
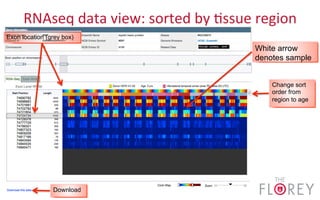
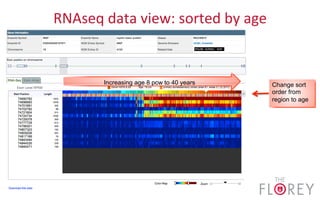
The document provides an overview of the UCSC Genome Browser, focusing on its applications in neuroscience, such as data visualization and analysis using various genomic data types. It discusses the functionality of loading and navigating the browser, accessing ENCODE data, and the types of information available, including gene annotations and expression data. Additionally, it outlines guidelines for utilizing data, the history of ENCODE's data policies, and emphasizes the importance of proper citations when using these resources for research.




























































































































![Polymer [ बहुलक ] Chemistry Notes PDF - Irfanullah Mehar - JJ Sir Chemistry.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/polymerchemistrynotespdf-irfanullahmehar-jjsirchemistry-260210172118-3f9b37f7-thumbnail.jpg?width=640&height=640&fit=bounds)


